





-
丙硫氧嘧啶(propylthiouracil),为硫脲类化合物,它可通过抑制甲状腺内过氧化物酶系统,阻止酪氨酸的碘化及碘化酪氨酸的缩合,从而抑制甲状腺激素的合成,是临床常用的治疗甲亢的药物[1-3]。其口服吸收迅速,约1~1.5 h达血药浓度峰值;代谢快,24 h内约有35%的药物以原型和葡萄糖醛酸化物的形式从尿中排出,血浆半衰期约为1~3 h[4]。口服丙硫氧嘧啶个体差异较大,由于其血浆蛋白结合率高(约为80%),且存在肝毒性,因此对其进行血药浓度监测,能使药物应用更为安全合理[5-6]。进口丙硫氧嘧啶的价格昂贵,研发高质量、低价格的国产仿制药具有良好的经济效益和社会效益[7],开发高效稳定的丙硫氧嘧啶的血药浓度测定方法,可为仿制药一致性评价中的生物等效性试验(BE)提供依据。
目前,丙硫氧嘧啶的体内测定方法主要为高效液相色谱法(HPLC)[8-10],早期开发的液相测定方法,分析时间长,专属性和灵敏度均无法满足临床高通量测定的需求,因此,建立简单快速、灵敏准确的丙硫氧嘧啶血药浓度质谱分析方法(MS)具有重要意义。本文建立UPLC-MS/MS法测定人血浆中丙硫氧嘧啶的含量,可为临床治疗药物监测(TDM)和生物等效性试验(BE)提供方法学基础。
HTML
-
Agilent 1290 UPLC超高效液相色谱(安捷伦科技有限公司,美国),配有在线脱气机、二元泵、低温自动进样器和柱温箱;Agilent 6470 Triple Quad三重四极杆质谱(安捷伦科技有限公司,美国),配有AJS-ESI喷射流电喷雾离子源、MassHunter分析工作站;SECURA125-1CN型十万分之一电子天平、Arium mini超纯水仪(赛多利斯,德国);Fresco 21低温离心机(赛默飞,美国)。
-
对照品丙硫氧嘧啶(批号:100803-201503,纯度>99.4%)购自中国食品药品检定研究院,内标丙硫氧嘧啶-D5(批号:13-EQJ-150-1,纯度>98%),购自Toronto Research Chemicals;甲酸为色谱纯(ACS恩科化学,美国);甲醇和乙腈为色谱纯(霍尼韦尔,美国);水为超纯水。
1.1. 仪器
1.2. 试药
-
色谱柱为Agilent SB-C18(4.6 mm×150 mm,5 μm);流动相采用甲醇-0.1%甲酸水溶液(80∶20,V/V),等度洗脱;流速:1 ml/min,柱后分流比2∶3;柱温30 ℃;进样量5 μl,分析时间3min。
-
采用AJS-ESI源,正离子模式,离子源参数:干燥气温度350 ℃;干燥气流速10 L/min;雾化器压力40 psi;鞘气温度350 ℃;鞘气流速11 L/min;毛细管电压4 000 V。多反应监测(MRM)的参数:丙硫氧嘧啶m/z 171.1→112.1,碎片电压110 V,碰撞能量30 eV;丙硫氧嘧啶-D5(IS)m/z 176.1→117.0,碎片电压110 V,碰撞能量30 eV。丙硫氧嘧啶和氘代内标的保留时间均为1.9 min。
-
精密称取丙硫氧嘧啶对照品1.5mg,置于2 ml称量瓶中,用移液器(已校准)加入甲醇适量溶解,涡旋混匀,配成1.0 mg/ml的对照品储备液;取上述对照品储备液适量,用甲醇逐级稀释,配成浓度分别为200、500、1000、2 000、10 000、20 000、40 000、80 000、100 000 ng/ml的系列对照品溶液,置4 ℃冰箱保存,备用。
-
精密称取丙硫氧嘧啶-D5对照品1.3mg,置于2 ml称量瓶中,用移液器(已校准)加入甲醇适量溶解,涡旋混匀,配成1.0 mg/ml的内标储备液;取上述储备液适量,用乙腈(含5%甲酸)稀释400倍,得2 500 ng/ml的内标溶液,置于4 ℃冰箱保存,备用。
-
取空白血浆950 μl,精密加入“2.2.1”项下制备的丙硫氧嘧啶系列标准溶液50 μl,旋涡混匀,配成浓度分别为10、25、50、100、500、1 000、2 000、4 000、5 000 ng/ml的标准含药血浆。同法制备4个浓度的质控样品(QC),分别为10、25、500、4 000 ng/ml,待用。
-
血浆样品先置于室温下解冻,取100 μl血浆,依次加入200 μl内标溶液,200 μl乙腈(含5%甲酸),涡旋1min,于4 ℃下13000×g高速离心10 min,取100 μl上清液于进样瓶中,进行UPLC-MS/MS分析。
-
通过比较6个不同健康志愿者的空白血浆样品、质控样品和给药后实际样品的色谱图来评估。分别取空白血浆、质控样品(QC-L)和受试者给药后的血样各100 μl,按“2.3”项下血浆样品前处理方法操作,进样,获得样品的色谱图(图1),包括空白样品色谱图、质控样品色谱图和实际样品色谱图。结果显示,空白血浆中的内源性成分不干扰待测物和内标出峰,选择性良好。
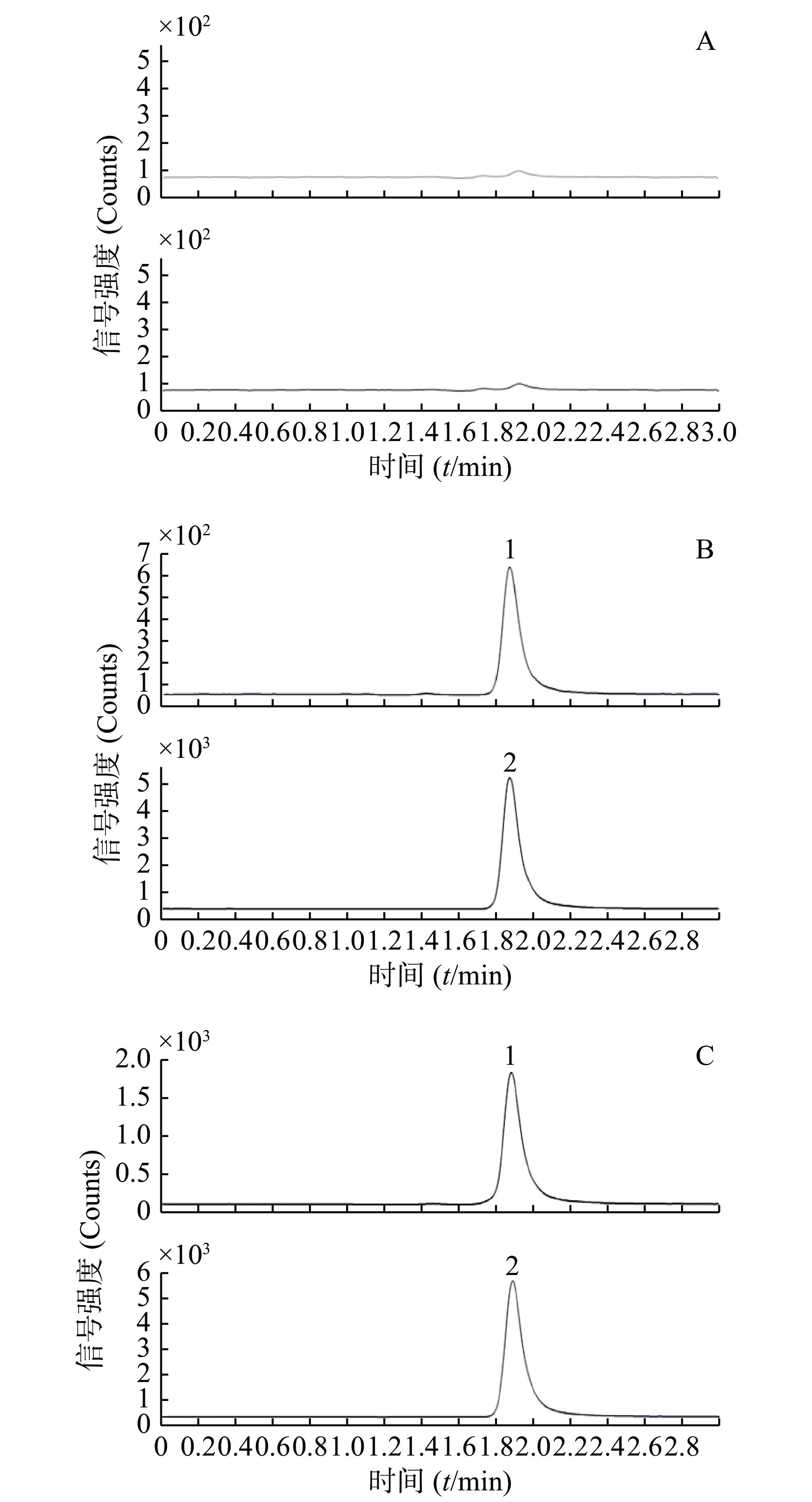
-
取“2.2.3”项下制备的标准含药血浆样品100 μl,按“2.3”项下血浆样品前处理方法操作,进样分析,记录色谱图。以待测物血浆浓度为横坐标(X),待测物与内标峰面积比为纵坐标(Y),进行回归,使用1/X 加权,求得回归方程: Y=0.000 4X−0.000 2(r=0.999 3)。结果表明,血浆中的丙硫氧嘧啶在10~5 000 ng/ml范围内线性关系良好。以S/N>10确定,定量下限(LLOQ)10 ng/ml,为标准曲线最低点。
-
在LLQQ(10 ng/ml)、QC-L(25 ng/ml)、QC-M(500 ng/ml)、QC-H(4 000 ng/ml)4个浓度下,通过批内和批间分别考察。按“2.2.3”项下制备丙硫氧嘧啶4个浓度质控样品,每个浓度平行5份,按“2.3”项下血浆样品前处理操作,进样分析。并于每天制备4个浓度质控样本各5份,进样分析,连续3 d,计算批内、批间的精密度(RSD)及准确度(RE),批内和批间的RSD<10%, RE<±10%,结果见表1。
浓度(ng/ml) 批内(n=5) 批间(n=15) 精密度(RSD/%) 准确度 (RE/%) 精密度(RSD/%) 准确度(RE/%) 10 3.40 −0.55 8.38 −0.88 25 8.35 −5.59 9.53 1.63 500 7.61 −1.00 6.21 −3.39 4 000 2.90 −2.79 4.71 −5.95 -
取6位健康志愿者的空白血浆,在相当于QC-L(25 ng/ml)和QC-H(4000 ng/ml)的2个浓度下来考察。先按“2.3”项下血浆样品前处理方法制备空白基质上清,然后加入丙硫氧嘧啶及内标的标准溶液,以使其终浓度与处理后QC-L和QC-H一致;同时制备相同浓度不含基质的待测物和内标的乙腈溶液,进样分析。通过峰面积分别计算待测物和内标的基质因子,进一步得到经内标归一化的基质因子。不同基质下,2个浓度基质效应的变异系数均小于5%,符合方法学要求,结果见表2。
浓度(ng/ml) 待测物
基质因子(%)内标
基质因子(%)归一化
基质因子(%)变异系数(%) 25 25.94 23.99 108.18 1.78 4 000 21.91 21.92 100.62 0.66 -
取单一来源的空白血浆,在相当于QC-L(25 ng/ml)、QC-M(500 ng/ml)、QC-H(4 000 ng/ml)的3个浓度下来考察。先按“2.3”项下血浆样品前处理方法制备空白基质上清,然后加入丙硫氧嘧啶及内标的标准溶液,以使其终浓度与处理后QC-L、QC-M、QC-H一致;同时按“2.2.3”和“2.3”项下制备和处理3个浓度的QC样品,每个浓度平行操作5份,分别进样分析。通过峰面积计算待测物的提取回收率,平均回收率为101.60%~113.56%,结果见表3。
被分析物 浓度(ng/ml) 回收率(%) RSD(%) 丙硫氧嘧啶 25 101.60 5.30 500 113.56 1.72 4 000 108.82 6.26 -
首先通过新鲜配制丙硫氧嘧啶和内标的储备液1.0 mg/ml,考察对照品储备液于4 ℃下放置30 d的稳定性,结果显示,RE<±10%,稳定性良好。然后在QC-L(25 ng/ml)、QC-H(4 000 ng/ml)2个浓度下,分别考察4种条件下丙硫氧嘧啶的稳定性:室温放置4 h、自动进样器(4 ℃)放置24 h,冻融循环3次,以及−80 ℃下冻存30 d,每个浓度平行操作5份。按“2.3”项下血浆样品前处理操作,进样分析,将丙硫氧嘧啶和内标峰面积的比值代入随行标准曲线求得实测浓度,计算RE以及RSD,结果(见表4)显示稳定性良好。
浓度(ng/ml) 室温放置4h 4 ℃放置24h 冻融循环3次 −80 ℃冻存30 d RE(%) RSD(%) RE(%) RSD(%) RE(%) RSD(%) RE(%) RSD(%) 25 2.82 6.58 1.99 6.00 4.67 4.85 8.17 5.59 4 000 −4.65 6.24 −7.70 4.51 4.82 3.90 3.73 1.93 -
通过在定量上限(ULOQ)5 000 ng/ml完成测定之后立即测定空白样品来评估。结果显示,分析物保留时间处峰面积小于LLOQ的20%,IS保留时间处的峰面积小于实际IS的5%。该方法几乎无残留,不影响测定。
-
通过使用空白基质将定量上限(ULOQ)10倍(50 000 ng/ml)和50倍(250 000 ng/ml)浓度的样品稀释至定量范围(10~5 000 ng/ml)来评估,每个浓度平行操作5次。结果表明,RSD和RE均低于15%,样品稀释不影响测定的精密度和准确度。
2.1. 分析条件
2.1.1. 色谱条件
2.1.2. 质谱条件
2.2. 溶液的配制
2.2.1. 丙硫氧嘧啶标准溶液
2.2.2. 内标标准溶液
2.2.3. 标准含药血浆及质控样品溶液
2.3. 血浆样品前处理
2.4. 血浆样品分析方法验证
2.4.1. 选择性
2.4.2. 标准曲线与定量下限
2.4.3. 精密度和准确度
2.4.4. 基质效应
2.4.5. 提取回收率
2.4.6. 稳定性
2.4.7. 残留
2.4.8. 稀释可靠性
-
常用的样品前处理方法为蛋白沉淀和液液萃取,优先采用简单的蛋白沉淀法。沉淀剂考察了甲醇和乙腈,结果发现乙腈沉淀更完全,在同位素内标下,归一化的基质效应更低,添加5%甲酸后,待测物峰形更好,因此选择乙腈(含5%甲酸)为沉淀剂。进一步考察了血浆与沉淀剂的比例(1∶3、1∶4、1∶5,V/V),结果发现,比例为1∶4时,提取回收率最高,最终选定为前处理方法。
-
首先考察了甲醇-水和乙腈-水体系,结果发现,虽然乙腈-水体系的洗脱能力更强,但甲醇-水体系下峰形更佳,血浆中内源性成分与主峰分离完全,基线噪音小。在水相中添加0.1%的甲酸,可以显著提高丙硫氧嘧啶的质谱响应,检测灵敏度令人满意,也可进一步改善峰拖尾。柱后采用了2∶3分流,实际进入质谱的流速约为0.4 ml/min,既保障了色谱柱的最佳流速(1 ml/min),又保证了ESI源的离子化效率,结果稳定可靠。
-
丙硫氧嘧啶在正离子模式下的响应明显优于负离子模式,因此确定采用正离子模式检测。采用Optimizer质谱参数优化程序依次对雾化室参数(鞘气温度、鞘气流速、干燥气温度、干燥气流速、雾化器压力、毛细管电压)以及MRM参数(母离子、子离子、碎片电压、碰撞能)进行调节优化,最终依据丙硫氧嘧啶和内标的检测灵敏度,确定了“2.2.2”项下最佳的质谱参数。得益于采用的同位素内标,丙硫氧嘧啶和氘代内标出峰稳定,虽保留时间同为1.9 min,但并没有离子串扰影响,而且归一化的基质效应结果满意。
综上,本文建立了快速简便、灵敏准确的测定人血浆中丙硫氧嘧啶含量的UPLC-MS/MS方法。样品采用简单蛋白沉淀法处理,LLOQ为10 ng/ml,基质效应低,回收率高,每个样本的分析时间3 min,每天可分析超过400样本,满足高通量测定需要。本研究为丙硫氧嘧啶的治疗药物监测(TDM)和生物等效性试验(BE)提供了方法学基础。
 DownLoad:
DownLoad: