



-
肝纤维化是肝脏对慢性肝损伤的过度修复反应,是一个动态可逆的过程。阻断肝纤维化持续进展是防治肝病向肝硬化甚至肝细胞癌发展的重要策略。目前研究认为,肝星状细胞(HSCs)转化为活化的肌成纤维细胞(MFBs)是细胞外基质 (ECM)的主要来源,也是肝纤维化的主要驱动力[1]。除此之外,大量研究发现,肝巨噬细胞在肝纤维化过程中同样发挥重要作用。
-
肝巨噬细胞是肝脏中一个高度异质的非实质细胞群,占肝脏细胞的10%~15%,主要由枯否细胞 (KCs) 和单核细胞衍生的各种浸润巨噬细胞 (MoMFs) 组成,具有显著异质性。巨噬细胞的异质性以多种来源、多种细胞类型特异性标志物和极化表型为特征。此外,肝巨噬细胞具有显著的可塑性,能够快速响应组织环境的变化,呈现不同的细胞表型。肝巨噬细胞被认为是肝脏抵御病原体的第一道防线,参与包括炎症反应、纤维化形成以及纤维化消退在内的肝纤维化所有阶段[2]。因此,肝内巨噬细胞是肝纤维化的核心调控者,肝纤维化治疗的重要靶细胞。
-
KCs是体内最大的常驻巨噬细胞群,包括卵黄囊来源的KCs和骨髓来源的KCs。KCs可以识别内源性细胞碎片和外源性病原体,感知肝脏损伤进而发生活化。因此,KCs对于肝脏稳态的维持、免疫反应的启动和肝损伤的恢复至关重要。
-
MoMFs充当免疫反应协调者并补充巨噬细胞群以维持肝脏中的稳态。在小鼠体内,MoMFs分为两个亚群:Ly6Chi和Ly6Clo单核细胞。Ly6Chi单核细胞参与炎症反应并产生促炎作用,而Ly6Clo单核细胞则具有组织修复功能。
-
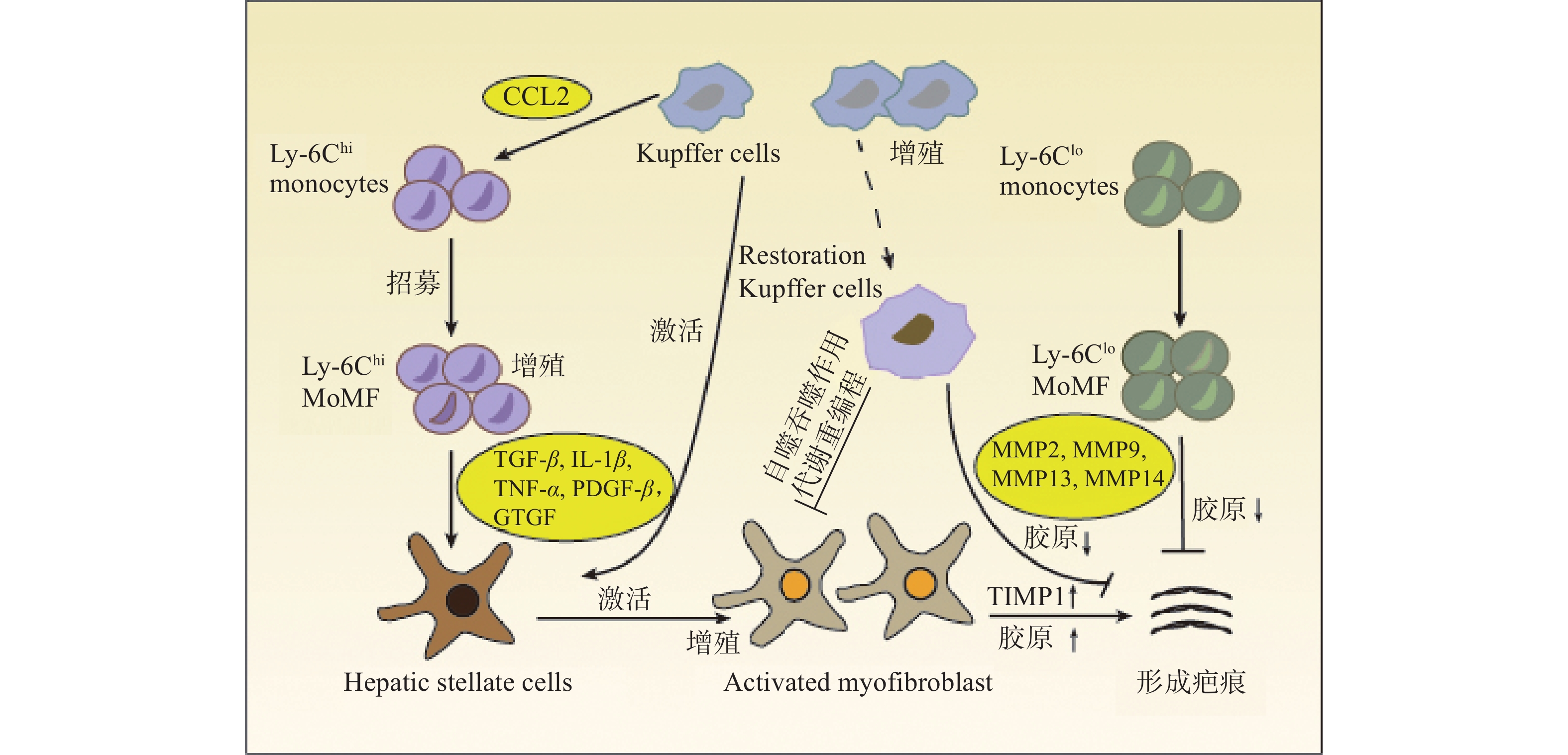
在肝损伤早期,损伤或死亡的肝细胞能够释放损伤相关分子模式 (DAMP),并与肝巨噬细胞相互作用,从而促进肝巨噬细胞的活化、极化和募集。活化的肝巨噬细胞能够分泌大量促炎因子和促纤维化细胞因子,参与炎症反应并促进HSCs的活化和纤维化应答[3](图1)。
-
HSCs位于肝细胞和肝窦内皮细胞之间的窦周间隙,在正常和受损的肝组织中具有不同的生理功能。正常的生理条件下,HSCs被称为静息的HSCs (qHSCs),负责储存肝脏中的维生素A。当肝损伤后,qHSCs被炎症介质激活,进而分化为 MFBs,分泌大量的ECM并产生纤维瘢痕,导致肝纤维化[4]。肝巨噬细胞能够与HSCs相互作用,重塑免疫微环境和ECM,该相互作用对于肝脏炎症应答和纤维化应答至关重要。
肝巨噬细胞能够分泌与HSCs活化有关的各种细胞因子和趋化因子,如转化生长因子β (TGF-β),肿瘤坏死因子α (TNF-α),白细胞介素1β (IL-1β) 和趋化因子配体2 (CCL2) 等。肝巨噬细胞产生的IL-1β和TNF-α能够激活NF-κB信号通路,增强MFBs的增殖[5]。激活素A (ACTA) 是TGF-β超家族成员之一,能够促进KCs中TNF-α和TGF-β1的表达,诱导HSCs转化为促纤维化表型,提高α-平滑肌肌动蛋白 (α-SMA) 表达水平和迁移能力[6]。此外,肝脏在受到丙型肝炎病毒 (HCV) 感染后,肝巨噬细胞能够分泌趋化因子配体5 (CCL5) ,引起HSCs中炎症小体和纤维化标志物α-SMA与TGF-β的激活[7]。
除细胞因子和趋化因子外,肝巨噬细胞还能够产生其他与HSCs串扰的分子。例如,MoMFs能够通过分泌颗粒蛋白激活qHSCs,促进其转化为MFBs,引起ECM的积累和肝纤维化的形成[8]。HMGB1是一种响应组织损伤而产生的DAMP,主要由肝细胞和KCs分泌,研究表明,HMGB1能够与HSCs中的晚期糖基化终末产物受体(RAGE)结合,激活pMEK1/2、pERK1/2和p-c-Jun通路,增加Ⅰ型胶原蛋白的沉积[9]。
此外,HSCs也可作用于肝巨噬细胞,影响其功能。紫藤凝集素阳性Mac-2结合蛋白 (WFA+-M2BP) 是一种肝纤维化的血清指示剂,可由HSCs分泌。据报道,WFA+-M2BP能够促进KCs表达M2BP,而KCs分泌的M2BP反过来又会进一步加强HSCs的激活。由此说明,在肝纤维化的发展过程中,巨噬细胞和HSCs之间存在正反馈调节[10]。
-
巨噬细胞具有显著的异质性,可以在不同的组织微环境中极化成不同的表型,从而发挥不同作用。通常,巨噬细胞极化的表型可以分为经典活化 (M1) 型和选择活化 (M2) 型。M2型可以进一步分为M2a、M2b、M2c和M2d亚型。
在被脂多糖 (LPS) 和干扰素-γ(IFN-γ) 刺激后,M1型巨噬细胞发生活化,分泌大量促炎细胞因子,如IL-1β、TNF-α、诱导型一氧化氮合酶 (iNOS) 等,从而发挥促炎作用。因此,M1型巨噬细胞在肝损伤起始阶段起重要作用,能够释放促炎因子,加剧炎症反应并促进肌成纤维细胞增殖,最终导致肝纤维化[11]。
M2型巨噬细胞的极化主要由白细胞介素4 (IL-4) 和白细胞介素13 (IL-13)等细胞因子诱导发生,能够分泌抗炎因子,如白细胞介素10 (IL-10) 、TGF-β、精氨酸酶1 (Arg-1) 等,具有抑制炎症、促进组织重塑、预防寄生虫感染以及调节免疫等生物学功能。在肝纤维化的发展过程中,M2型巨噬细胞产生抗炎作用,能够促进伤口的愈合和再生。然而,当肝损伤持续存在时,M2型巨噬细胞释放的TGF-β、血小板衍生生长因子 (PDGF) 、血管内皮生长因子 (VEGF) 等生长因子,会促进MFBs的增殖和活化,加重肝纤维化[12]。
调节巨噬细胞极化可以发挥抗肝纤维化作用。脯氨酸-丝氨酸-苏氨酸-磷酸酶相互作用蛋白2 (PSTPIP2) 能够调节STAT1和NF-κB信号通路,抑制M1型巨噬细胞极化,改善肝脏炎症和肝纤维化[13]。白细胞介素22 (IL-22) 能够调节STAT3、Erk、Akt信号通路的传导,促进巨噬细胞从M1型转变为M2型,从而减缓肝纤维化的进展[14]。
此外,肝巨噬细胞和其他细胞群之间的相互作用对肝巨噬细胞的表型转换也至关重要。自然杀伤 (NK) 细胞在调节巨噬细胞极化方面发挥关键作用,研究发现,DX5+NKp46+ NK细胞能够产生IFN-γ促进M1巨噬细胞极化,在预防非酒精性脂肪性肝炎 (NASH) 进展为肝纤维化方面发挥重要作用[15]。中性粒细胞 (PMN) 能够促进巨噬细胞转变为具有肝脏修复功能的巨噬细胞,有助于肝脏炎症和肝纤维化的自发消退[16]。
-
肝巨噬细胞浸润与慢性炎症和肝纤维化密切相关。在趋化因子和相应受体的介导下,巨噬细胞能够募集到损伤部位,参与炎症和肝纤维化的发展。例如,Ly6Chi MoMFs能够依赖趋化因子受体2 (CCR2) 募集到肝损伤区域,发挥促炎和促纤维化作用。微蛋白 (PSMP) 是一种新型趋化因子,能够促进炎性巨噬细胞的浸润,分泌更多的促炎细胞因子,加剧肝纤维化应答[17]。骨髓细胞上表达的触发受体1 (TREM-1) 可促进KCs的募集和浸润以及促炎细胞因子的产生[18]。
阻碍肝巨噬细胞的浸润与募集有助于肝纤维化的消退。配对免疫球蛋白样2型受体α (PILRα) 是一种抑制性受体,主要在骨髓细胞中表达,在炎症过程中能够抑制肝巨噬细胞与PMN的浸润。研究证实,PILRα能够调节整合素信号传导,阻碍巨噬细胞迁移到受损的肝组织,从而减轻肝脏炎症并缓解肝纤维化[19]。
此外,研究还发现一种新的TREM2+ CD9+瘢痕相关巨噬细胞 (SAMacs) 亚群,其来源于MOMFs,在肝纤维化过程中表现出促纤维化表型[20],可能成为未来肝纤维化治疗的重要靶细胞。
-
在肝纤维化进展中,肝细胞能够分泌一系列DAMP和细胞外囊泡 (EVs) ,可与肝巨噬细胞相互作用,诱导巨噬细胞转变为促炎表型。线粒体DNA (mtDNA) 是一种内源性DAMP,能够激活先天免疫反应[21]。肝细胞衍生的mtDNA能够激活NF-κB信号通路,诱导KCs分泌TNF-α和白细胞介素6 (IL-6) ,引起肝脏炎症和纤维化应答[22]。M1型巨噬细胞分泌的EVs能够激活肝细胞中NLRP3炎性体信号通路,而应激的肝细胞可分泌含有微小RNA-192-50 (miR-192-5p) 和血清CD40L配体 (CD40L) 的EVs,促进M1型巨噬细胞极化[23]。因此,肝细胞和巨噬细胞之间通过释放DAMP和EVs相互作用,促进了肝内炎症反应与肝纤维化进展。
-
细胞代谢重编程是细胞为满足能量需求,通过改变代谢模式促进细胞增殖和生长的机制,包括糖代谢、脂代谢、氨基酸代谢等。
肝巨噬细胞的代谢重编程与巨噬细胞极化紧密联系,影响肝纤维化的进展和消退[24]。c-Rel是NF-κB转录因子家族成员之一,参与巨噬细胞代谢重编程。研究发现,c-Rel能够与6-磷酸果糖激酶-2的启动子结合,诱导巨噬细胞极化和HSCs活化,从而加重炎症反应与肝纤维化[25]。此外,肝脏中铁的代谢失调也与晚期肝纤维化有关。肝内铁的积累能够激活MiT/TFE转录因子,促进M1型巨噬细胞的活化,加重肝纤维化[26]。
膜联蛋白A5是膜联蛋白家族的成员之一,能够与M2型丙酮酸激酶(PKM2)相互作用,将肝巨噬细胞中的糖酵解转换为氧化磷酸化,促进巨噬细胞从M1型转换到M2型,从而改善炎症和肝纤维化[27]。因此,调节肝巨噬细胞的免疫代谢是肝纤维化的潜在治疗策略。
-
自噬是将机体中异常表达的蛋白质和受损的细胞器转移到溶酶体中进行降解,对细胞稳态的维持、细胞存活、分化和生长至关重要。大量研究证实,巨噬细胞自噬对肝脏具有保护作用。例如,KCs的自噬能够抑制细胞活性氧 (ROS) 介导的白细胞介素1α (IL-1α) 和IL-1β的分泌,从而缓解肝脏炎症和纤维化[28]。日本血吸虫卵抗原 (SEA) 诱导的巨噬细胞自噬能够抑制肝脏病理的发展[29]。白细胞介素7 (IL-7) 能够通过激活AMP活化蛋白激酶 (AMPK) 抑制SEA诱导的巨噬细胞自噬,促进炎症细胞对肝脏的浸润,增强MFBs活性,从而加重SEA感染引起的肝纤维化[30]。此外,LC3相关的吞噬作用 (LAP) 是一种非典型的自噬形式,能够将Ly6Chi MoMFs变为Ly6Clo MoMFs。研究表明,LAP可抑制全身炎症,发挥抗肝纤维化作用[31]。
吞噬作用是细胞摄取较大固体颗粒或大分子复合体的过程。肝巨噬细胞能够通过吞噬和清除肝脏中死亡的细胞调节肝脏炎症和纤维化。在肝损伤中,巨噬细胞能够吞噬坏死的肝细胞,诱导Wnt3a的表达并激活Wnt通路,从而促进肝再生。肝脏巨噬细胞的吞噬作用,减弱了受损肝细胞中线粒体衍生的DAMP的释放,从而抑制肝脏瘢痕的形成[32]。因此,调节肝巨噬细胞的自噬和吞噬功能可成为一种新的抗肝纤维化策略,值得进一步研究。
-
除了KCs和MoMFs,其他浸润性巨噬细胞群也与肝纤维化有关。SOCS蛋白是巨噬细胞炎症活性的调节因子,在肝纤维化期间,脾巨噬细胞能够通过上调肝巨噬细胞中的SOCS3信号传导来促进CCL2的分泌,从而促进循环单核细胞的浸润,加剧肝纤维化的发展[33]。
-
肝纤维化的发展与多种细胞和分子机制相关。除肝巨噬细胞外,HSCs、肝细胞、肝窦内皮细胞、胆管细胞和脾细胞也参与了肝纤维化的发展,这些细胞之间的相互作用,能够调控细胞内信号传导,从而影响肝纤维化进展和消退。此外,肝内肝窦的形成和重塑是肝纤维化的关键特征,抑制血管生成也能够减缓肝纤维化的进展。
-
肝巨噬细胞在肝损伤、肝纤维化进展和消退中发挥双重作用。目前研究证实,多种药物能够通过调控肝巨噬细胞的功能发挥抗肝纤维化的作用。因此,基于巨噬细胞在肝纤维化中的作用,人们开发了相关的趋化因子抑制剂、细胞通路拮抗剂,期望为肝纤维化提供新的治疗策略。
-
调节巨噬细胞极化可以治疗肝纤维化。例如,槲皮素能够调控Notch1通路,抑制M1型巨噬细胞极化,缓解炎症反应,从而抑制肝纤维化进展[34];壳寡糖能够通过调控JAK2/STAT1和JAK1/STAT6信号通路,抑制巨噬细胞极化为M1型,增加M2型巨噬细胞数量,从而发挥抗肝纤维化作用[35]。此外,研究还发现,在肝纤维化期间,脾切除能够激活ERK1/2信号通路,促进MOMFs转换为抗炎的Ly6CloMOMFs,从而减轻肝脏炎症和肝纤维化应答[36]。
-
抑制巨噬细胞募集和浸润有助于肝纤维化的消退。姜黄素能够通过抑制KCs的激活减少趋化因子分泌,降低Ly6ChiMOMFs的浸润,从而缓解肝纤维化[37]。鉴于CCL2/CCR2和CCL5/CCR5信号通路在巨噬细胞募集中的关键作用,人们研发出了相关的趋化因子受体拮抗剂,如CCR2拮抗剂RS102895、CCR2/CCR5双拮抗剂 (CVC) 。研究发现,在酒精性肝纤维化模型中,CVC能够明显抑制体内巨噬细胞的募集,展现出较好的的抗纤维化活性[38]。
-
巨噬细胞自噬是一种针对慢性肝损伤和纤维化的保护机制,通过诱导巨噬细胞自噬能够治疗肝纤维化。MJN110是一种单酰基甘油脂肪酶 (MAGL) 抑制剂,在CCl4和BDL诱导的肝纤维化模型中,MJN110的干预能够促使巨噬细胞自噬通量和自噬体生物合成增加、减少肝巨噬细胞数量,从而减缓肝纤维化进展,促进肝纤维化消退[39]。
-
大多数基于肝巨噬细胞的疗法仅在肝纤维化的动物模型中进行了评估,而相关的临床研究数据较少。CVC是CCR2和CCR5双重拮抗剂,两项临床实验数据显示,CVC在伴有纤维化的NASH中具有显著的抗纤维化作用,并且耐受性良好[40]。此外,有研究人员在人体上进行了自体巨噬细胞治疗的安全性评估实验,结果表明,该疗法在肝硬化患者中是安全可行的,这为未来研究其在肝硬化和其他纤维化疾病中的疗效提供了依据[41]。
-
肝纤维化是由各种病因所致慢性肝损伤的修复反应,其特征是ECM在肝内的过度沉积。鉴于肝巨噬细胞在调节肝纤维化反应中的关键作用,人们开发出了针对肝巨噬细胞治疗肝纤维化的新策略。基于抑制KCs活化的靶向疗法已被研究,这些疗法主要通过抑制细胞内炎症信号通路,如NF-κB、ASK1、JNK和p38等信号通路,从而治疗肝纤维化[42]。Loomba等人开发了Selonsertib,一种ASK1信号通路的抑制剂,研究证实,Selonsertib对肝细胞代谢和巨噬细胞活化有影响。在一项随机2期试验中,Selonsertib能够降低NASH和肝纤维化患者的肝脏中胶原蛋白含量和小叶炎症程度,并且能够改善肝细胞凋亡和坏死[43]。此外,肝纤维化治疗的重点是减少MoMFs向肝脏的募集。MoMFs向受损肝脏的募集依赖于活化的肝细胞分泌的几种趋化因子,如趋化因子配体1 (CCL1) ,CCL2,CCL5[44]。因此,调节趋化因子的信号传导也是一种治疗策略,这些疗法主要包括针对趋化因子或受体的单克隆抗体、阻止趋化因子结合的受体拮抗剂、适体分子和阻断趋化因子诱导的细胞内信号传导的小分子抑制剂等[5]。研究发现,使用CCR2敲除能够减弱小鼠MoMFs募集,抑制MFBs活化并减轻肝纤维化[45]。此外,MoMFs可分为导致肝脏损伤的Ly6Chi MOMFs和具有肝脏修复功能的Ly6Clo MOMFs。因此,另一种潜在的策略是通过将Ly6Chi MOMFs转换为Ly6Clo MOMFs来恢复正常的肝功能。研究证实,在CCl4诱导的肝纤维化模型和MCD饮食诱导的NASH模型中,CCL2抑制剂mNOX-E36能够抑制Ly6Chi MOMFs的早期流入,同时能够将Ly6Chi MOMFs转换为Ly6Clo MOMFs,促进肝纤维化的消退[46]。
尽管肝巨噬细胞在肝纤维化发病机制中的作用机制和相关治疗策略已经取得了突破性进展,然而,通过巨噬细胞靶向肝纤维化疗法仍然存在局限性。需要解决的问题如下:肝巨噬细胞的这些表型其临床意义是什么,是否有可能对肝巨噬细胞进行基因改造以解决肝纤维化,如何达到只靶向致病表型而不破坏正常的生理表型?此外,大多数关于肝巨噬细胞的作用和潜在机制的研究都是在啮齿动物模型中进行的,由于啮齿动物和人类之间的肝巨噬细胞存有差异,这些发现与人类的相关性仍需要进一步研究。
Research progress on hepatic macrophages and liver fibrosis
doi: 10.12206/j.issn.2097-2024.202209030
- Received Date: 2022-09-12
- Rev Recd Date: 2023-10-22
- Available Online: 2024-04-24
- Publish Date: 2024-04-25
-
Key words:
- hepatic macrophages /
- liver fibrosis /
- phenotypic switch
Abstract: Liver fibrosis is a repair response to chronic liver injury caused by various etiologies. Its continuous progression can develop into liver cirrhosis or even hepatocellular carcinoma, eventually leading to liver failure. Currently, there is no effective treatment for liver fibrosis. Hepatic macrophages play a key role in intrahepatic inflammatory response, progression and resolution of fibrosis, and have emerged as an important therapeutic target for anti-hepatic fibrosis. The function of hepatic macrophages in the process of liver fibrosis was mainly reviewed and the mode of action of hepatic macrophages from various aspects was discussed to provide ideas for the treatment of liver fibrosis.
| Citation: | JIANG Haiyan, ZHONG Fangfang, ZHANG Junping. Research progress on hepatic macrophages and liver fibrosis[J]. Journal of Pharmaceutical Practice and Service, 2024, 42(4): 135-140. doi: 10.12206/j.issn.2097-2024.202209030

|
 DownLoad:
DownLoad: