





-
我国是仿制药大国,但是仿制药的研发水平参差不齐,许多药企的研发工作尚不规范。工艺研究可分为两个阶段:实验室小试研究和中试放大研究。其中小试研究的目的主要是确定关键质量属性、关键工艺参数以及参数区间等,保证工艺路线以及最终成品质量的稳定性,是对原始工艺的优化及补充[1]。
六西格玛管理是由摩托罗拉公司倡导的一种企业流程设计、改进和优化的管理模式,近几年在许多领域得到了广泛应用。DMAIC模型是实施六西格玛管理的一套基于数据的管理工具与操作方法。通过持续运行的DMAIC模型可以实现质量或管理等的持续改进,具体包括定义(define)、测量(measure)、分析(analyze)、改进(improve)、控制(control)5个阶段[2]。笔者在此基础上结合仿制药小试研究的特点,探究如何以DMAIC模型来改进仿制药小试研究项目管理流程,并通过某原料药企业案例验证其可行性,以期为我国仿制药小试研究提供借鉴,提高仿制药的研发水平。
HTML
-
DMAIC模型强调以实际问题为改善活动的输入,运用统计过程控制等工具采集问题及过程相关的数据,充分利用数据波动来寻找问题存在的主要原因及改进措施,通过相应的措施来验证改进措施的有效性,实现降低问题缺陷率及过程变异[3]。本文根据仿制药小试研发的特点,建立适合仿制药小试研究的DMAIC模型,各阶段的主要工作以及方法如图1所示。

目前,质量源于设计(QbD)是国内外比较流行的用于药品开发的系统方法,是动态药品生产管理规范(cGMP)的基本组成部分[4]。而DMAIC模型是对QbD设计的一种补充与完善,使研发过程能够更加规范,符合我国GMP和注册申报的要求,也促进后续向大生产的转换。DMAIC模型更加强调项目管理以及实验流程的科学性和真实性,具体异同点总结见表1。
DMAIC模型 QbD 不同点 组织方式 在界定环节进行团队以及项目管理 从实验角度出发,不涉及此项 CQAs和CPPs确定方式 使用统计方法,如帕累托图以及线性分析 多使用风险评估的方式,如失败模式和效应分析[5] 测量系统 强调测量系统的科学准确,
确保实验符合GMP要求从实验角度出发,不涉及此项 控制策略的制定与确认 除界定设计空间外,还涉及 “人、机、料、法、
环”;重复实验确定控制措施的有效性控制策略主要是对参数进行控制,并不考虑大
生产的实际操作可行性;无重复实验要求相同点 设计思路一致。均以预先设定的目标产品质量概况,确定产品的关键质量属性(CQAs)和关键工艺参数(CPPs),研究CPPs
对产品关键质量属性的影响并建立工艺设计空间,进而提出能满足产品性能且工艺稳健的控制策略[5]
-
仿制药的研究以药学研究为主,主要进行工艺以及质量对比研究,具体流程包括小试、中试以及工艺验证等[6]。小试直接关系着药品注册工作进程和实验室向生产转移的顺利程度。其研究水平越高,向大生产工艺过渡的成功率越高[7]。但是由于小试研究属于前置性研究,追求效益的企业管理者往往忽视其作用而导致小试研究水平的落后,甚至编造虚假数据以求通过注册申报。我国仿制药企业在小试研究过程中主要存在以下3个问题。
-
小试研究是一项系统工作,需要多个部门的配合。但在我国实际操作过程中,小试研究工作往往由研发部门独立进行,缺少生产部门、设备部门、质量部门的参与[7]。小试研究缺乏完整的计划安排,往往会导致任务衔接不畅甚至出错。同时,部门配合不紧密、沟通存在障碍也会拖延小试的进度,不仅造成时间和资金上的浪费,还易导致小试研究的不规范。由于缺乏经验,许多研发人员对试验的安排较为随意,导致试验的有效性和可靠性存疑。
-
在仿制药研发过程中,研发与生产脱节成为许多企业最为困扰的问题,例如,小试摸索出的工艺条件与企业现有大生产条件不符,因此无法进行大生产操作,或者关键工艺参数范围不易控制等。这种情况的根源在于:研发人员对于实际生产的认知和重视程度低,没有用生产的眼光和GMP指导小试研究工作[8]。
-
在2010年版《药品生产质量管理规范》中明确指出药品的设计与研发应体现本规范的要求[9]。《药品数据管理规范》(征求意见稿)中描述数据可靠性管理适用于药品研发、生产、流通、上市后监测与评价等产品生命周期中全部活动的数据管理[10]。然而,我国多数制药企业的研发部门缺乏GMP意识,小试研究工作往往不符合药品GMP的要求,这就导致研发阶段提供的资料可能从形式、内容上无法满足申报的要求,例如,记录的缺失、不完整等等。近几年,许多制药企业的生产现场检查发现文件管理不到位或记录不规范的问题 [11-12]。
2.1. 开发统筹安排差
2.2. 研究与生产衔接差
2.3. 质量管理不规范
-
某化学仿制原料药A为非甾体激素类药,主要用于过敏性休克、支气管哮喘等。产品的质量直接关系到疗效,但产品的质量受多方面影响,例如,物料供应商所供应材料质量,生产工艺过程中反应温度、pH值等参数的变动以及操作工艺执行的规范性,都有可能引起产品的质量缺陷。为保证产品质量的可靠性与真实性以及能完成原料药申报注册,该企业研发人员对生产工艺进行了小试研究,确定关键参数以及设计空间,并优化初始工艺,以生产出符合国家药典以及制剂要求的原料药。该原料药的生产工艺主要包括胺化、还原、拆分、游离、干燥、包装,本次DMAIC模型应用于还原步骤的工艺参数优化。
-
在定义阶段,需进行项目选题、团队建设以及确定改进目标Y。该项目主题确定为还原工艺参数优化,能够生产质量达标且稳定的还原产物。团队以研发部门为主,指定专门的仪器以及QC人员辅助检测,注册部负责整个项目的工作协调。在该环节最为关键的是界定改进问题的范围,明确什么问题需要改善并重点关注。该化学原料药A以胺化产物作为原料,通过氢化还原成中间体2。如果中间体2的质量存在显著问题,会导致后续反应杂质多或收率低,对最后成品的有关物质和收率等项目具有关键影响。结合该中间体对成品的影响,分析了可能存在的4个品质关键点CTQ(critical to quality),各类CTQ的测量方法、标准及依据如表2所示。通过帕累托图(图2)发现,该中间体有关物质项最大单一杂质不合格的数量最多,占比最高,是影响合成产物质量的关键因素。因此,本次小试改进目标Y确定为降低最大单杂至0.5%以下并且工艺稳定。该项目的测量方法为2015年版《中国药典》的成品的高效液相法,可以通过该方法判断工艺路线中杂质的清除情况并能够获得较为准确的数据。该企业在此阶段进行了人员组织安排以及项目进度管理,使小试能够在有计划、有安排、有专人负责的情况下进行,小试研究效率较以往有大幅提升。另外,通过帕累托图发现CQAs更具说服力。
品质关键点 测量方法 标准 依据 外观 目测 类白色结晶性
粉末文献资料 氢化液纯度 高效液相法 纯度≥99.5% 自拟① 碱纯度 高效液相法 ≥98.0% 自拟① 消旋碱有关物质 高效液相法 最大单杂≤0.5% 自拟① 收率 数学计算 ≥75% 实际生产情况 注:“①”表示标准是根据最终药品质量指标反向推算以及工艺重现时该项的最佳数值确定。 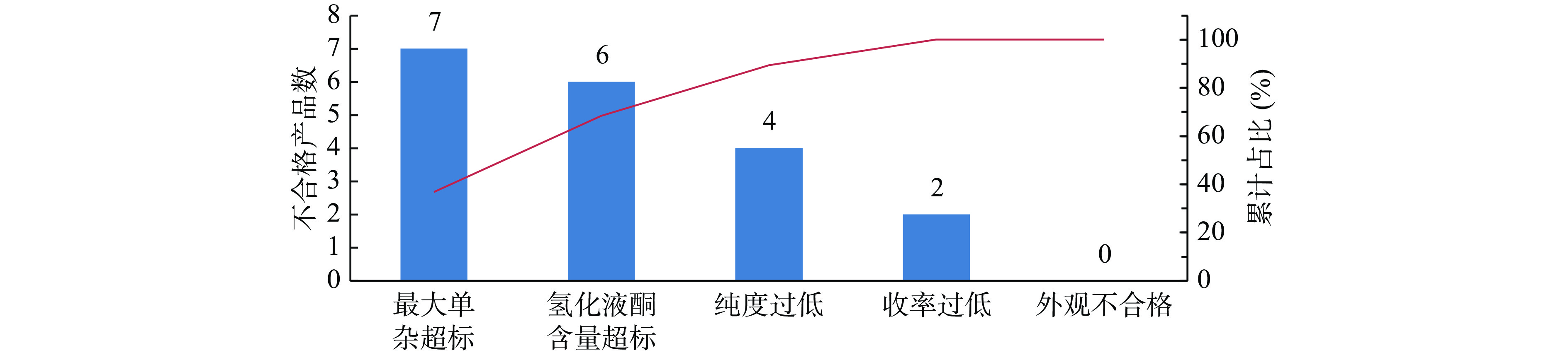
-
在该阶段的主要任务是将项目转换成一个统计的问题,分为3步:确定影响目标Y的因素,验证Y的测量系统并进行数据的搜集整理。首先,查阅文献并结合工作经验,通过头脑风暴法认为还原反应温度、反应液pH、碱化游离温度、游离pH这4项是引入杂质的重要因素。另外,根据产品的质量要求对目标Y划分等级,确定最大单杂>0.5%为不合格,≤0.5%为合格。为了确保测试所得数据的准确和可靠,测量系统需处于稳定状态,不受外部因素影响。真实、规范、完整的实验记录是保证药品测量系统真实可靠的基础。因此,本试验工作所使用的物料、仪器设备和采用的试验条件、试验方法、操作步骤、试验过程、试验结果、数据以及结论等均进行记录,确保研究轨迹清楚、可追溯并且可重复。编制小试检验原始记录表,对检验试剂、检验仪器、检验步骤、检验方法、检验结果等如实记录。在该阶段,研发试验人员也需编写小试方案。本案例中,研发人员针对4项影响因子进行单因素试验,确定试验条件、试验步骤等,由QA进行审核,经研发部主管批准。小试方案批准后,研发试验人员负责数据的搜集和整理。其中,单因素试验的因素为上述4项,每个因素选3个水平进行设计,每个试验条件平行试验2次,共有24批样本。试验设计如表3所示。在质量部门的配合下,研发人员根据QC部门的检验结果进行统计,在保证试验数据真实、完整的情况下,完成了数据搜集工作。通过DMAIC模型对测量系统的规范,实验数据得到真实、完整的记录,并且单因素试验设计能够较为科学地对比出关键工艺参数以及设计区间。该流程在符合QbD设计的同时,确保了研发数据的真实性和可溯性。
序号 还原反应温度
(t/℃)反应液pH值 碱化游离
温度(t/℃)游离pH值 1 XX±5 X~X XX±5 ≈8 2 XX±5 X~X XX±5 ≈9 3 XX±5 X XX±5 ≈10 -
该阶段的主要任务是对已收集到的数据,通过统计分析等,找到影响因素与目标之间的关系,聚焦问题核心并最终找出关键因素以及范围。在本案例中,通过对小试试验数据的分析发现,还原反应温度和反应液pH值是影响目标Y值的关键因素。具体分析如表4所示。通过原始数据以及电子化数据的相互印证,为后续的中试以及注册申报提供清晰的数据支撑,至此,基本获得了注册要求的工艺开发优化的过程资料。
序号 工艺参数 范围 依据 是否关键 1 还原反应
温度XX~XX℃ 温度高时一未知杂质含量
较高,且难清除。在该
范围内,综合收率和纯
度都在较高水平是 2 反应液
pH值≈X 随着反应液pH值的提高,
氢化液中主要杂质迅速
降低,关系式为y = 7.65e-
1.215x(r= 0.987 8)。
pH为X时,杂质含量最小是 3 碱化游离
温度XX~XX℃ 该范围内纯度和
收率基本稳定否 4 游离pH值 X~X 该范围内纯度和
收率基本稳定否 -
改进阶段是DMAIC模型的核心阶段,改进实施的效果决定了DMAIC模型的应用水平。在本项目中,改进措施为按照试验结果所确定的参数条件,从“人、机、料、法、环”五大角度对参数进行控制。在后续试验中,按照上述条件生产了5批中间体2,检验发现有关物质一项最大单杂均≤0.5%,满足小试预期目标并且工艺稳定。由此得出结论:改善对策的效果是显著的,满足本次小试研究的目标。并且验证改进措施的有效性降低了研发的不确定性和风险。
-
当上述步骤逐步实施后,项目便进入控制阶段。该阶段的主要任务是为后续建立标准化的文件,包括项目报告以及指导规范等。在本案例中,小试项目组首先通过编写小试总结报告,将上述改进措施文件化并与生产部门共同起草生产工艺规程以及岗位操作法等。在后续中试及实际生产过程中,对每个班次的操作人员进行培训和稽核,确保所有的改善对策都有被执行,操作人员依照生产工艺规程作业,所有设备依照文件进行确认以及校验。该步骤巩固了本次小试研究的成果,方便注册文件的编写。与生产部门共同起草工艺规程等能够保证后续大生产的有效衔接,在工业化生产前解决了许多矛盾。
3.1. 定义阶段
3.2. 测量阶段
3.3. 分析阶段
3.4. 改进阶段
3.5. 控制阶段
-
DMAIC模型和QbD的研究理念是一致的,能够充分满足注册申报以及商业化生产的要求,提高注册和药政管理的灵活性。与QbD设计相比,DMAIC模型有如下优势:①在问题阶段就提出需进行团队建设,建立项目管理的整体框架,有效解决小试安排统筹差的问题。②利用统计而非风险评估判断CQAs和CPPs,避免了研发人员的主观判断,提升了小试研究的科学性。③在测量阶段要求测量系统是科学可靠的,其检验方法、检验结果都有迹可循。④重复改进措施实验,确保改进措施的有效性,降低意外风险。⑤文件化改进措施以及“人、机、料、法、环”的应用促使小试研究人员用生产的眼光看待小试研究,将小试研究结果与实际生产相结合。通过本案例保证了该模型的小试研究是在QbD理念的基础上,系统、有逻辑地、在符合GMP要求的情况下进行。但基于DMAIC模型的特性,它只适用于原始工艺较成熟,小试目的为优化并控制工艺稳定的情况。不过DMAIC模型作为一种先进的管理集成方式,在仿制药小试研究中仍可大有作为。
 DownLoad:
DownLoad: