



-
临床药物研究需要严格、标准、规范的数据管理[1],众多文献阐述了统计分析前临床试验数据核查的相关问题及改进措施[1-4],然而,对于统计分析所得数据的核查却鲜有报道。本文以生物等效性(BE)研究为例,介绍统计分析数据核查的要点,包括:试验分组的随机数字表、药代动力学(PK)主要参数以及BE的分析计算数据和结果在相应的软件中是否能够重现、与原统计分析报告是否一致。同时,统计专业人员对核查提出的问题进行敏感性分析,并出具相关报告。
-
本文所使用的数据来自我部于2019-2021年间所接受的18项BE研究统计分析数据核查的结果,所测试的药物分别为抗病毒、抗菌药物以及治疗心脑血管疾病、糖尿病等疾病的药物。
-
随机指利用SAS软件中的随机化功能,事先给出种子数,进行随机化分组。该方法简便易行、可重复、符合随机化要求[5]。通过SAS系统的“PROC PLAN SEED=种子数”过程实现两组等比例随机化。例如,将001~010这10个数随机分配为A、B两组,种子数设定为20200506,则生成的结果A组为002、003、006、008和009,B组为001、004、005、007和010。核查时,只要采用试验时设定的种子数,则通过SAS运行得到的A组和B组的数据不变。用同样的随机种子,通过SAS程序运行,能够得出相同的随机表。由此证明研究对象进入试验组和对照组机会均等。
-
BE指药学等效制剂或可替换药物在相同的试验条件下,给予相同的剂量,其活性成分吸收速度和程度的差异无统计学意义[6]。BE研究给药后,通过测量不同时间点的生物样本(全血、血浆、血清等)药物浓度,得出药物浓度-时间曲线,经过计算得出血药浓度-时间曲线下面积(AUC)、药物达峰浓度(cmax)、达峰时间(tmax)(图1)等PK参数后,再通过统计学分析比较,判断两种制剂是否生物等效。
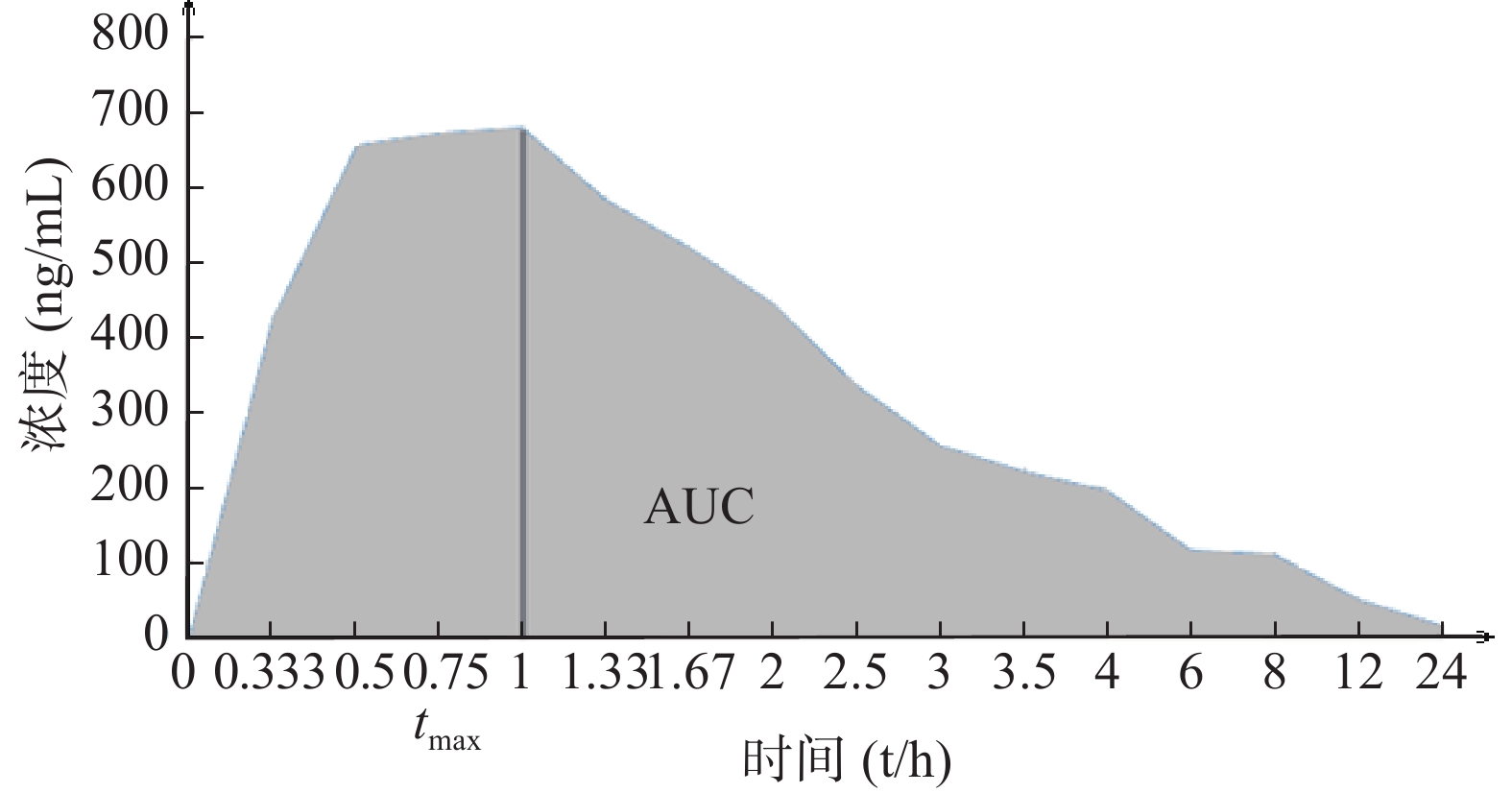
目前,BE评价方法是置信区间法。当主要PK参数AUC和cmax的几何均值比值的90%置信区间在80%~125%内时,受试制剂吸收的速度和程度与参比制剂相当,视为生物等效[7]。
核查时,首先需要核对申办方提供的统计分析前的原始数据。统计专业人员向核查人员展示数据传输协议(由申办方提供);然后,根据原始样本浓度数据,现场使用WinNonlin 8.1软件处理各组血药浓度测定数据,采用非房室模型计算出主要PK参数,包括AUC0-t、AUC0-∞、cmax,其他数据则使用SAS 9.4软件分析。如果受试制剂与参比制剂的AUC0-t、AUC0-∞和cmax几何均数比值的90%置信区间均落在80%~125%范围内,则两种制剂生物等效;否则,两种制剂不存在生物等效。将核查所得结果和结论与原统计分析报告核对,检查一致性。
-
18项BE研究的分组随机数字表均可重现;18项研究均符合生物等效的判定标准,且BE评价结果与原统计分析报告一致;其中有13项研究的PK参数与原统计分析报告相同,其余5项研究的PK参数则有差异,见“2.2”项。
-
有12项研究存在个别样本采样时间偏差,其中,2项研究被要求补充敏感性分析,3项研究的分析数据集被要求进行受试者数据的重新纳入或剔除后,也进行了敏感性分析。
上述5项研究敏感性分析的结果显示:PK参数AUC0-t、AUC0-∞和cmax的几何均值比值的90%置信区间虽在数值上有所变化,但仍然在80%~125%范围内,维持原统计分析报告的BE评价。
-
BE试验通常采用随机交叉等试验设计,随机和盲法因其有效控制偏倚而成为BE试验注册和核查的关键内容[8]。由于随机化即随机分配的优点,随机对照试验被广泛认为是评价新的药物、新的医疗器械或新的治疗方法疗效的最佳设计[9],是评价医学新疗法的金标准[10]。
在过去的二十年里,许多第一代专利药品的专利和上市许可的到期,导致了仿制药的兴起。进行BE研究被认为是确定仿制药与专利药具有相同的有效性和安全性的关键[7]。BE研究在化学药物仿制药的申请、新药评价以及已上市药物的变更申请中,具有不可替代的作用[6, 8, 11]。
-
在药物临床试验方案中,试验期间各个样本采集的时间点有严格规定。由于操作技术、环境及其它因素的影响,可能导致样本采集的时间与计划采样时间有所偏差、且超过了方案允许的偏差范围,这种情况视为超窗[12]。超窗会否影响统计分析的结果,需要通过敏感性分析予以证明。
敏感性分析指通过改变方法、模型、未测量的变量值等考查结果的改变程度,以确定评估方法的稳健性。敏感性分析结果与主要分析结果一致,表明主要分析的结果稳健,反之亦然[13]。对BE研究而言,考查的关键便是生物等效这一评估结果对PK参数的变化是否敏感。
核查发现,有2项BE研究分别存在个别受试者采血时间超窗问题。例如,在某研究中,某受试者计划采血时间为给药后5 min,方案规定的允许偏差范围为30s之内,但实际采血时间为5min32s,即超窗2s。在原统计分析报告中,PK参数按照计划采血时间加2s,即5min2s计算。核查人员要求按照实际采血时间重新计算主要PK参数,即按照5min32s计算,并做敏感性分析,与原统计分析结果进行比较,考查所得结论的一致性。
由此可见,对样本采集的环节进行严格、科学的管理非常重要。另外,无论采样时间是否存在偏差甚至超窗,应尽可能获取所有数据,真实记录采样时间,并做出敏感性分析报告。
-
临床试验有效性分析应涵盖随机化分组后的所有受试者,而不仅限于实际完成的受试者数据。按照这种意向性治疗原则所做的分析是最好的分析。
BE研究的统计分析集除全分析集(FAS)和安全数据集(SS)外,最主要的数据集为PK参数集(PKPS)和BE集(BES)[14],BES是推断受试制剂和参比制剂是否生物等效的数据集。
在撰写统计报告时,如果剔除了某受试者数据,或者有受试者出现种种事故但没有被剔除,核查时可能被要求重新纳入或剔除受试者的数据,并进行敏感性分析,以考查是否对最终结果造成影响。例如,某BE试验对照组某受试者第二周期无给药后3.50 h、3.75 h、4.00 h、4.25 h、4.50 h及以后共13个时间段的血药浓度数据,原报告将该受试者纳入PKPS与BES,在核查时,将该受试者第二周期剔除PKPS与BES;或者,原报告将类似受试者剔除PKPS与BES,核查时又将该受试者重新纳入PKPS与BES。
需要注意的是,BE研究通常样本量相对较小,受各种原因数据剔除造成数据缺失的影响相对较大,可能对BE统计分析结果的稳健性带来挑战。因此,BE研究须严格质量管理,事先没有规定的不做剔除处理[11]。
-
统计分析数据是临床研究结果的呈现,是撰写统计分析报告和临床研究报告的依据。为确保临床研究最终结果和结论没有争议,国家药品监督管理局核查中心对药物临床研究的统计分析数据进行核查非常必要。
本文从统计学角度介绍了BE研究统计分析数据现场核查的主要内容,并对相关统计方法、核查发现的问题、原因和对策进行逐一分析和讨论。考虑到BE研究通常样本量较小,采样时间出现偏差、剔除或纳入数据可能影响统计结果的稳健性,因此,敏感性分析在BE研究中尤为重要,用于评估主要分析结果和结论的稳健性。敏感性分析和主要分析结果一致,则补充、巩固和加强研究结论,进一步证实试验药物的有效性和安全性[15-17]。本文建议,BE研究在统计分析计划的制定与统计分析报告的撰写中,对核查的内容、可能涉及敏感性分析的相关问题,特别是敏感性数据集PKPS和BES的调整,应予以充分考虑,以便在数据分析阶段采用多种敏感性分析方法,综合考虑结果的稳健性,为评估不同制剂临床治疗的可替换性提供扎实的研究数据。
Data verification and sensitivity analysis of statistical analysis in bioequivalence studies
doi: 10.12206/j.issn.2097-2024.202212040
- Received Date: 2022-12-20
- Rev Recd Date: 2023-05-30
- Publish Date: 2023-06-25
-
Key words:
- bioequivalence studies /
- statistical analysis /
- data verification /
- sensitivity analysis
Abstract:
| Citation: | LI Linhe. Data verification and sensitivity analysis of statistical analysis in bioequivalence studies[J]. Journal of Pharmaceutical Practice and Service, 2023, 41(6): 377-379. doi: 10.12206/j.issn.2097-2024.202212040

|
 DownLoad:
DownLoad: