

 下载:
下载:

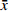
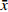
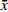
 下载:
下载:

-
玄膝荣筋散是由蚂蚁、青风藤、老鹳草、川芎、全蝎、重楼等十三味中药组成的复方制剂,其中黄芪为君药,白术、桂枝为臣药,具有滋补肝肾,养肝荣筋,消热利湿,祛风止痛。用于早、中、晚期风湿、寒型类风湿性关节炎。由于该标准中仅包含一个川芎薄层色谱鉴别和蚂蚁、黄芪的显微鉴别,且无定量测定方法,不能全面控制制剂质量。根据全军医疗机构制剂标准提高课题要求,在原有质量标准基础上,增加了川芎、穿山龙的薄层鉴别,同时采用高效液相色谱法,测定玄膝荣筋散中所含桂皮醛的含量,该法灵敏度高、专属性好、操作简便、重现性好。
-
LC-20AD岛津高效液相色谱仪(日本); 紫外检测器。
-
玄膝荣筋散(批号:140601、131226、140701,规格:250g/袋),由南京政治学院门诊部提供;桂皮醛(对照品,批号:110710-201418,中国食品药品检定研究院);乙腈为色谱纯,水为超纯水,其他试剂均为分析纯。
-
取本品5 g,加乙醚20 ml,超声处理15 min,滤过,滤液低温挥去乙醚,残渣加乙酸乙酯1 ml使溶解,作为供试品溶液。取川芎对照药材0.5 g,同法制成对照药材溶液。取缺川芎的阴性样品,同法制备阴性对照液。吸取上述3种溶液各10 μl,分别点于同一硅胶G薄层板上,以正己烷-乙酸乙酯 (9∶1)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上显相同颜色的斑点,阴性试验样品未见干扰,见图1。
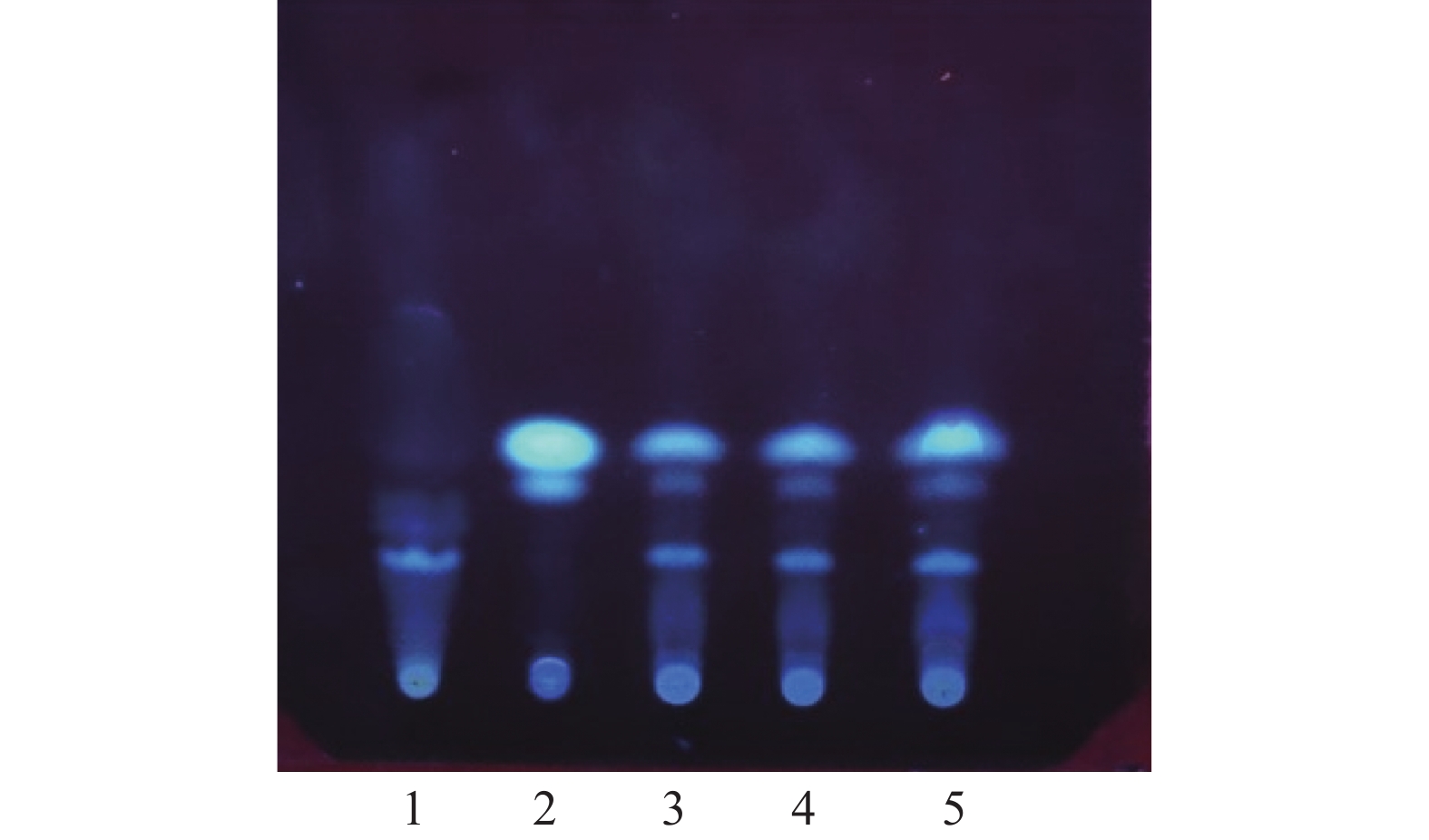
图 1 川芎薄层色谱图
-
取本品粉末15 g,加甲醇50 ml,加热回流1 h,滤过,滤液蒸干,残渣加甲醇1 ml使溶解,作为供试品溶液。另取穿山龙对照药材0.5 g,加甲醇25 ml,同法制成对照药材溶液。取缺穿山龙的阴性样品,同法制备阴性对照液。吸取上述3种溶液各5 μl,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水 (13∶7∶2)的下层溶液为展开剂,展开,取出,晾干,喷以10 %硫酸乙醇溶液,在105 ℃加热至斑点显色清晰,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上显相同颜色的斑点,阴性试验样品未见干扰,见图2。
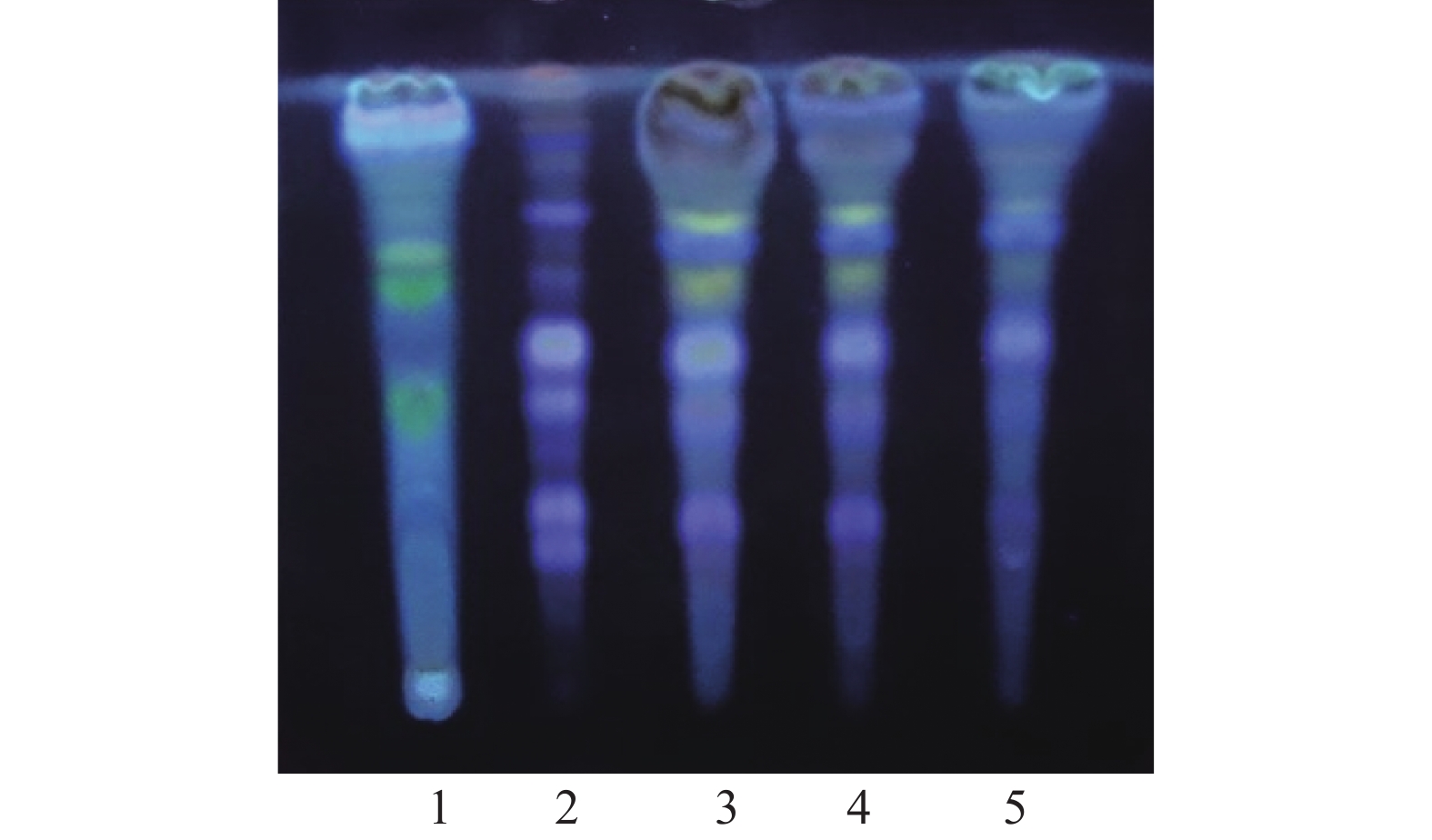
图 2 穿山龙薄层色谱图
-
色谱柱:KR100-5C18 (250mm×4.6mm ;5μm, E14878);流动相:乙腈-水(35∶65);流速:1.0 ml/min;检测波长290 nm;进样体积:10 μl。在此条件下,样品中桂皮醛与基线达到良好分离,见图3。
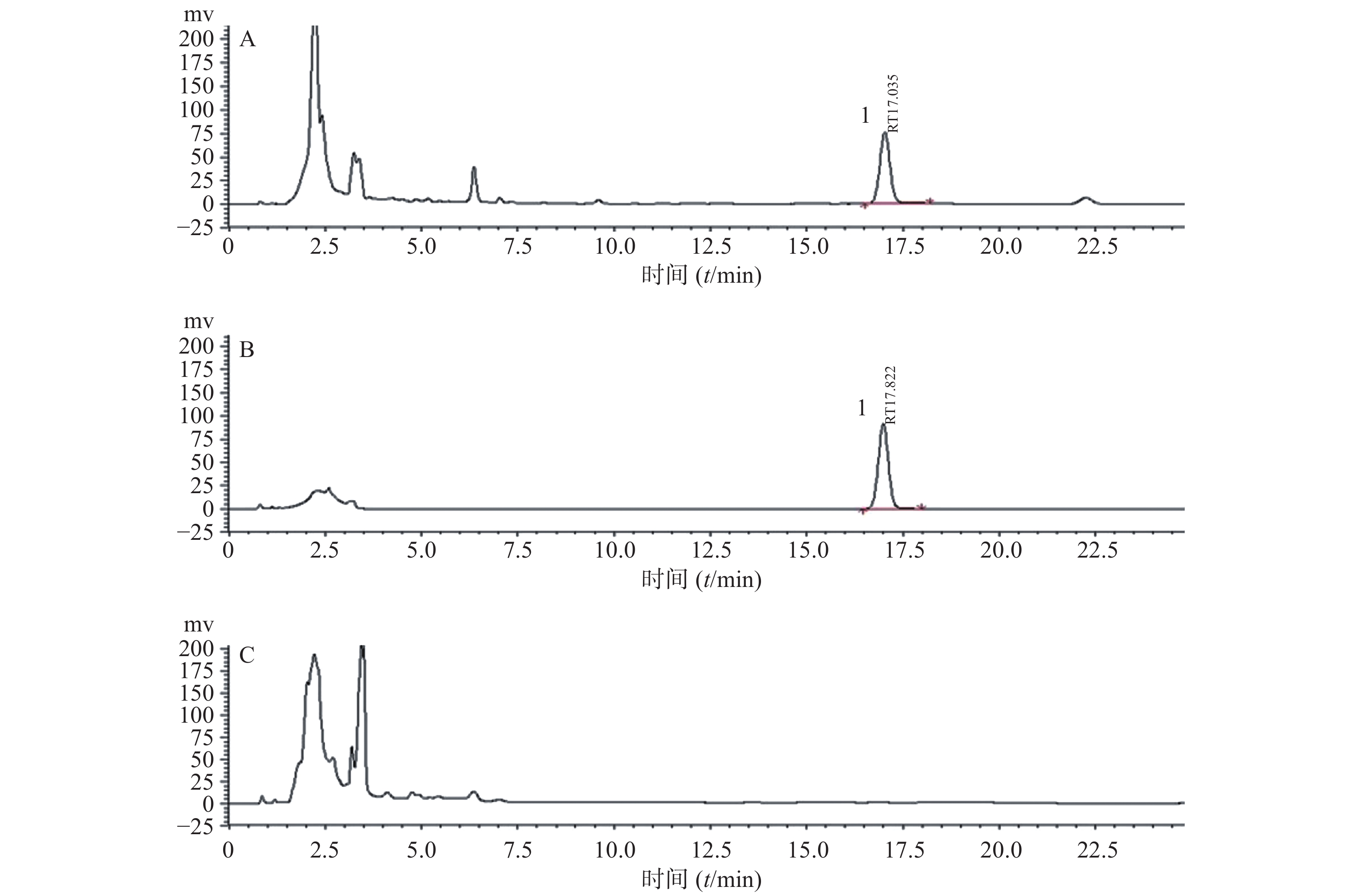
图 3 专属性试验HPLC图谱
-
取桂皮醛对照品适量,精密称定,加甲醇制成每1 ml含桂皮醛 15 µg的溶液,即得。
-
取样品(过四号筛)粉末约1 g,精密称定,置于具塞锥形瓶中,精密加入80 %乙醇25 ml,称定重量,超声处理(功率250 W,频率40 kHz)20 min,放冷,再称定重量,用 80 %乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。
-
按处方量制备缺桂枝的阴性样品,按供试品溶液方法制备阴性对照溶液。
-
取对照品溶液、供试品溶液、阴性对照溶液,按“2.2.1”项下色谱条件进样分析,阴性对照液中色谱峰对测定无干扰(见图3)。
-
取桂皮醛对照品适量,加甲醇溶解并制成每1 ml含0.0326 mg溶液,作为对照品溶液5,将溶液5分别稀释为0.00489、0.008150、0.0163、0.02445 mg/ml四种浓度,作为对照品溶液1~4。精密吸取上述对照品溶液1~5各10μl,注入液相色谱仪,按“2.2.1”项下色谱条件测定峰面积,以进样量(X)为横坐标,峰面积(Y)为纵坐标,绘制标准曲线,得回归方程为:Y=10181123.15X+6639.54,r=1.00,结果表明:桂皮醛在0.0489~0.3260 µg/ml范围内有良好的线性关系。
-
精密吸取“2.2.3”项下供试品溶液,在上述色谱条件下,连续进样6次。结果峰面积的RSD为1.07%(n=6),表明仪器精密度良好。
-
取同一批号的玄膝荣筋散(批号140601)按“2.2.3”项下制备方法平行制备6份供试品溶液,按上述色谱条件,测得桂皮醛峰面积RSD为1.12%,结果表明此法重复性良好。
-
取同一批号的玄膝荣筋散(批号140601)样品,精密称定,照上述含量测定方法,精密吸取10 µl, 分别在0、2、4、8、12、24 h进样,测定一次。结果桂皮醛峰面积的RSD为0.95%,表明供试品溶液在24 h内稳定性良好。
-
取同一批号的玄膝荣筋散(批号140601)0.5 g,精密称定,分别精密加入桂皮醛(0.00815 mg/ml)对照品溶液25 ml,按“2.2.3”项下制备方法平行制备6份供试品溶液,按“2.2.1”项下色谱条件测定,结果见表1。
表 1 桂皮醛加样回收率试验测定结果
称样量(g) 相当桂皮醛量(mg) 加入量(mg) 实测量(mg) 回收率(%) 平均回收率(%) RSD(%) 0.5078 0.1737 0.2038 0.3717 97.15 0.5045 0.1725 0.2038 0.3708 97.30 0.5064 0.1732 0.2038 0.3644 93.82 95.69 1.78 0.5018 0.1716 0.2038 0.3637 94.26 0.5051 0.1727 0.2038 0.3650 94.36 0.5006 0.1712 0.2038 0.3694 97.25 -
取3批样品,按上述供试品溶液制备方法处理,进样,计算桂皮醛的含量,见表2。
表 2 玄膝荣筋散中桂皮醛含量测定结果(n=3)
批号 含量(mg/g) 140601 0.3422 140701 0.3121 131226 0.2945 -
桂皮醛为桂枝药材的主成分,经紫外吸收光谱扫描(200~400 nm),在291 nm处有最大吸收,故确定检测波长为290 nm。
-
参照2020版《中国药典》[1] 中桂皮醛含量测定的方法,分别取样品2.0251、1.0530、0.5169 g,加入不同浓度的乙醇、甲醇等,超声处理20、30、40 min,按照正文的含量测定项下方法进行测定。结果表明,最终选择取样量为1g,选用80%乙醇为溶剂,在超声30 min后提取较完全。
-
建立该制剂中川芎、穿山龙的薄层色谱鉴别方法,拟增加黄芪、甘草的薄层实验中,因存在阴性试验样品有干扰的问题,故未列入正文。对TLC方法,分别从温度(10 °C和35 °C)、相对湿度(42%和88%)以及不同硅胶板(国产和进口)等方面进行考察。在各试验条件下,鉴别结果不受温度、相对湿度和不同硅胶板等条件改变的影响。说明本文所建立的薄层色谱方法作为定性鉴定方法具有很强的适用性。
本实验运用高效液相色谱法建立了桂皮醛含量测定的方法,经过试验验证,该法灵敏度高、专属性好、操作简便、重现性好,可作为该制剂生产过程中的质量控制及检验手段,更好把控药品疗效与临床用药安全。
Study on quality standard for Xuanxi Rongjin powder
-
摘要:
目的 修订玄膝荣筋散的定性和定量测定方法。 方法 采用薄层色谱(TLC)法对川芎、穿山龙进行定性鉴别,HPLC法测定制剂中桂皮醛的含量。采用KR100-5C18 色谱柱(250mm×4.6mm,5μm),流动相为乙腈-水(35∶65),检测波长为290 nm。 结果 TLC法能定性鉴别川芎、穿山龙。桂皮醛在0.0489~0.3260µg/ml范围内线性关系良好(r=1.00)。平均加样回收率为95.71%(RSD=1.78%)。 结论 该法灵敏度高、专属性好、操作简便、重现性好。 Abstract:Objective To revise the qualitative and quantitative determination methods of Xuanxi Rongjin powder. Methods TLC was used to qualitatively identify Chuanxiong and Chuanshanlong. The content of cinnamaldehyde in the preparation was determined by HPLC with KR100-5C18 column (250 mm×4.6 mm, 5μm). The mobile phase was acetonitrile-water (35:65) and the detection wavelength was 290 nm. Results TLC can qualitatively identify Chuanxiong and Chuanshanlong. Cinnamaldehyde has a good linear relationship in the range of 0.0489~0.3260 µg/ml (r=1.00), The average recovery was 95.71% (RSD=1.78%). Conclusion The method has high sensitivity, good specificity, simple operation and good reproducibility. -
Key words:
- Xuanxi Rongjin powder /
- quality standard /
- HPLC /
- TLC /
- cinnamaldehyde
-
替考拉宁(teicoplanin)是一种糖肽类抗生素,常用于治疗包括耐甲氧西林金黄色葡萄球菌(MRSA)在内的G+菌引起的感染[1]。它包括5个主要成分(TA 2-1、TA 2-2、TA 2-3、TA 2-4和TA 2-5)、1个水解成分(TA 3-1)和4个次要成分(RS-1~RS-4)[2]。其中,TA 2-2组分是活性最强的化合物,占各组分相对含量的40%以上[3-4],临床上常常通过测定TA 2-2的含量来监测替考拉宁的浓度[5-6]。目前,替考拉宁治疗药物监测主要以测定总浓度为主,但监测结果存在较大的个体差异。主要因为替考拉宁蛋白结合率较高(90 %~95%),在血液循环中主要与白蛋白(Alb)结合并处于动态平衡,未结合的游离部分在体内发挥药效[7]。健康状态下,人体白蛋白为正常水平(35~52 g/L),不同患者药物的血浆蛋白结合率变化不大;但在低蛋白血症(Alb<25 g/L)[8-10]、危重症、酸血症、肾衰竭、肝硬化、吸烟、多种药物合用等特殊情况下,患者常伴有白蛋白水平下降,当Alb<30 g/L时,白蛋白就会对替考拉宁游离浓度产生显著影响从而影响疗效[7, 10]。同时,游离药物水平升高导致肾脏排泄增加,肾毒性风险增加[11-12]。所以,针对上述白蛋白水平较低的患者,单纯用总浓度去评价替考拉宁的疗效和安全性是片面的,若同时测定替考拉宁游离浓度,将更加利于临床精准调整给药方案,以获得满意的治疗效果,减少或避免不良反应的发生。本研究旨在建立一种简便、快速、灵敏、准确的离心超滤结合超高效液相色谱串联质谱法(UPLC-MS/MS)用来测定替考拉宁游离浓度,为临床实现替考拉宁游离血药浓度监测提供依据。
1. 材料与方法
1.1 实验仪器
QTRAP 5500型三重四极杆/复合线性离子阱质谱仪(美国AB-SCIEX有限公司);30AD型超高效液相色谱仪(日本岛津有限公司);AL204型分析天平(梅特勒-托利多仪器上海有限公司);H1850R台式高速冷冻离心机(湖南湘仪实验室仪器开发有限公司);Vortex-5型涡旋混合机(江苏海门其林贝尔仪器制造有限公司);超纯水仪系统(默克密理博上海有限公司);Centrifree® 超过滤装置(截留分子量>30 000的物质)(默克密理博上海有限公司);DK-S24型电热恒温水浴器(上海精密试验设备有限公司)。
1.2 药品和试剂
替考拉宁对照品(中国食品药品检定研究院);甲酸、醋酸铵(色谱纯,Dikma科技有限公司);乙腈(色谱纯,Sigma-Aldrich公司)。
1.3 UPLC-MS/MS分析条件
1.3.1 UPLC分析条件
色谱柱:EndeavorsilTM C18柱 (50 mm×2.1 mm,1.8 µm);流动相A:0.02 mol/L醋酸铵溶液(含0.1%甲酸),流动相B:乙腈,梯度洗脱信息见表1;柱温:35 ℃;进样量:5 µl。
表 1 梯度洗脱信息时间(t/min) 流速(ml/min) A(%) B(%) 0.0 0.2 85 15 11.0 0.2 74 26 13.0 0.2 74 26 15.0 0.2 85 15 20.0 0.2 停止 停止 1.3.2 MS检测条件离子源参数:
喷雾针电压(ISV):5 500 V;碰撞气(CAD):medium;气帘气(CUR):20 psi;入口电压(EP):10 V;去簇电压(DP):78 V;碰撞能量(CE):20.74 V;碰撞池出口电压(CXP):13 V;离子源气体1(GS1):55 psi;离子源气体2(GS2):60 psi;离子源温度(TEM):550 ℃。检测模式:电喷雾离子化(ESI),选择性离子检测(SIM),正离子。多离子反应监测模式(MRM)离子对:替考拉宁TA2-2 m/z 940.7→316.2。扫描时间:200 ms。
1.4 工作液的配制
精密称取替考拉宁对照品2.0 mg置于100 ml容量瓶中,用超纯水溶解并稀释至刻度,得到浓度为20.0 μg/ml的储备液。分别取不同体积的替考拉宁储备液,用超纯水稀释成浓度为0.20、0.50、1.00、2.00、4.00、8.00、12.0、16.0 μg/ml的标准曲线工作液,以及0.50、3.00、12.0 μg/ml的质控工作液。
1.5 血浆样品的处理
取1 ml血浆样品置于Centrifree®超滤装置(截留分子量>30 000的物质)中,在37 ℃水浴中平衡30 min,然后在37 ℃,1 500×g下离心30 min,取样品超滤液直接进样。
1.6 血浆超滤液样品的配制
分别取“1.4”项下各浓度标准曲线工作液以及质控工作液100 μl,加入100 μl空白血浆超滤液,稀释成浓度为0.10、0.25、0.50、1.00、2.00、4.00、6.00、8.00 μg/ml的标准曲线血浆超滤液样品,以及浓度为0.25、1.50、6.00 μg/ml的质控点血浆超滤液样品。
2. 结果
2.1 专属性
取空白血浆超滤液,直接进样,得到色谱图A;空白血浆超滤液加替考拉宁对照品溶液配制成一定浓度(1.00 μg/ml),混合均匀后进样,得色谱图B;取使用替考拉宁的患者血浆,按“1.5”项下处理,进样后得色谱图C,考察方法的专属性。根据色谱图1中A、B、C,可以看出血浆中内源性物质没有干扰替考拉宁的检测,同时TA 2-2与TA 2-3完全分开,TA 2-2保留时间为11.57 min,可以准确定量TA 2-2。
2.2 线性关系考察
分别取“1.6”项下各浓度标准曲线血浆超滤液样品5 μl直接进样分析,以替考拉宁游离浓度为横坐标(X),峰面积为纵坐标(Y),得线性回归方程为Y=51 708.90X+1 397.10(r=0.999,n=8),结果表明,替考拉宁游离浓度在0.10~8.00 μg/ml范围内,与峰面积呈良好的线性关系,定量下限为0.10 μg/ml。
2.3 精密度与准确度
按照“1.6”项下质控点血浆超滤液样品配制方法,配制低、中、高浓度(0.25、1.50、6.00 μg/ml)的质控点血浆超滤液样品各6份,直接进样分析,计算批内精密度。按照相同的方法重复测定3次,计算批间精密度。结果见表2。如表2所示,在低、中、高浓度水平,样品的批内和批间RSD (%)均不超过7.00%,实测值与理论值相对误差在−5.00%~7.00%之间,精密度与准确度均符合生物样品测定要求。
表 2 批内和批间精密度与准确度(n=6,$ \bar x$ ±s)理论浓度
(μg/ml)实测浓度
(μg/ml)准确度
(%)精密度(%) 批内 批间 0.25 0.25±0.02 1.56 6.91 6.77 1.50 1.43±0.05 −4.52 2.53 3.83 6.00 5.95±0.20 −0.88 2.99 3.44 2.4 方法回收率
按照“1.6”项下质控点血浆超滤液样品配制方法,配制低、中、高浓度(0.25、1.50、6.00 μg/ml)的质控点血浆超滤液样品各6份,直接进样分析,按线性回归方程计算实测浓度,以实测浓度与理论浓度的比值计算方法回收率。表3结果显示,低、中、高浓度水平的回收率在93.8%~101%范围内,总回收率为97.9%,同时RSD (%)也符合生物样品分析要求。
表 3 替考拉宁的方法回收率(n=6,$ \bar x$ ±s)浓度水平
(μg/ml)回收率
(%)RSD
(%)总回收率
(%)总RSD
(%)0.25 101±7.00 6.91 97.9 5.50 1.50 93.8±2.37 2.53 6.00 98.5±2.96 3.01 2.5 基质效应
取来源不同的6个空白血浆样品,按照“1.6”项下质控点血浆超滤液样品配制方法,每个来源都分别配制成低、中、高浓度(0.25、1.50、6.00 μg/ml)的质控点血浆超滤液样品(每个浓度3份),直接进样分析,得峰面积A1。此外,用超纯水配制低、中、高浓度(0.25、1.50、6.00 μg/ml)的替考拉宁对照品溶液(每个浓度3份),直接进样分析,得峰面积A2。计算相应样品中替考拉宁的A1/A2值,即为基质因子(MF),考察基质效应。表4结果显示,低、中、高浓度水平所有样品的平均基质因子为0.97,接近1.00,且RSD (%)为2.12%,符合生物样品分析要求,说明基质效应不会影响分析方法的准确性。
表 4 替考拉宁在血浆超滤液中的基质效应(n=3,$ \bar x$ ±s)浓度水平(μg/ml) 基质因子 0.25 1.50 6.00 基质A 0.96±0.02 0.96±0.01 0.98±0.02 基质B 0.95±0.03 0.96±0.03 0.97±0.01 基质C 1.00±0.01 0.97±0.01 0.99±0.01 基质D 0.97±0.02 1.00±0.01 0.98±0.01 基质E 0.99±0.03 0.98±0.02 0.99±0.03 基质F 0.97±0.02 0.95±0.01 0.98±0.01 各浓度水平均值 0.97 0.97 0.98 各浓度水平RSD (%) 2.58 2.21 1.30 总均值 0.97 总RSD (%) 2.12 2.6 稳定性考察
2.6.1 血浆超滤液样品在室温条件下的稳定性
配制低、中、高浓度(0.25、1.50、6.00 μg/ml)的质控点血浆超滤液样品,每个浓度有6个重复样品,放置在室温10 h后与当日配制的标准曲线同批测定,考察替考拉宁血浆超滤液样品在室温下的稳定性。结果见表5,室温放置10 h后,样品实测浓度与理论浓度偏差在−4.00%~2.17%,RSD(%)不超过6.00%,在生物样品分析要求范围内,表明样品在室温下放置10 h后稳定性较好。
表 5 不同储存条件下替考拉宁样品的稳定性(n=6,$ \bar x$ ±s)储存条件 理论浓度(μg/ml) 实测浓度(μg/ml) RSD (%) 准确度(%) 室温10 h 0.25 0.24±0.01 5.52 −4.00 1.50 1.48±0.03 2.16 −1.33 6.00 6.13±0.12 2.09 2.17 −20 ℃ 15 d 0.25 0.22±0.01 4.76 −12.00 1.50 1.46±0.02 1.98 −2.67 6.00 5.79±0.13 2.23 −3.50 冻融3次 0.25 0.22±0.01 6.43 −12.00 1.50 1.44±0.05 3.67 −4.00 6.00 5.68±0.18 3.15 −5.33 2.6.2 血浆超滤液样品长期冷冻保存的稳定性
配制低、中、高浓度(0.25、1.50、6.00 μg/ml)的质控点血浆超滤液样品,每个浓度有6个重复的样品,在−20 ℃放置15 d后与测定当日配制的标准曲线同批进样分析,考察替考拉宁血浆样品−20 ℃下的稳定性。结果见表5,−20 ℃下储存15 d后,样品实测浓度与理论浓度偏差在−12.0%~−2.67%,RSD在5.00%以内,符合生物样品分析要求,表明样品在−20 ℃保存15 d后稳定性达标。
2.6.3 血浆超滤液样品的冻融稳定性
配制低、中、高浓度(0.25、1.50、6.00 μg/ml)的质控点血浆超滤液样品,每个浓度有6个重复的样品,放置在−20 ℃的冰箱内超过24 h,取出放置在室温条件下完全溶解后,再放置在−20 ℃的冰箱内,反复冻融3次,在第3次融化后与测定当日配制的标准曲线同批进样分析,以考察血浆样品3次冻融循环稳定性。结果见表5,反复冻融3次后,样品实测浓度与理论浓度偏差在−12.0%~−4.00%,RSD在6.50%以内,符合生物样品分析要求,表明样品冻融后稳定性仍较好。
3. 总结与讨论
近年来,血浆药物游离浓度的监测逐渐受到广泛关注。对于临床上提倡进行治疗药物监测,而蛋白结合率又高于80%的药物,建议监测药物游离浓度用以指导用药的剂量调整。对于高蛋白结合率的药物来说,白蛋白水平的改变可能会导致其药动学参数变异性增加[12]。因此,对于低蛋白血症(Alb<25 g/L)[8-10]、危重症、酸血症、肾衰竭、肝硬化、吸烟、多种药物合用等这类常伴有白蛋白水平降低的患者来说,替考拉宁的蛋白结合率具有一定的变异性[7, 13],即使获得相同的替考拉宁总浓度,体内真正发挥药效的游离替考拉宁浓度也可能不同。此时,应准确测定替考拉宁游离浓度来调整用药剂量,以防浓度低达不到治疗目标,或用药过量导致不良反应的发生。为实现对替考拉宁游离浓度的监测,本研究首次建立了超滤离心结合UPLC-MS/MS的方法。该方法与传统的高效液相色谱法相比,优点在于检测速度快,检测限较低,灵敏度较高,质谱检测可以排除患者所用的其他药物对替考拉宁的干扰,准确度较高。替考拉宁在水中易溶,在乙腈、甲醇中几乎不溶。因此,用超纯水作为储备液溶剂可使对照品完全溶解。替考拉宁成分复杂,为实现对各组分的有效分离,本研究选用Dikma公司的EndeavorsilTM C18柱,是通用型UPLC反相柱,具有独特的选择性和超高的分离性能,适合于高效、超快速分离分析,且峰形对称性好。在流动相选择的前期探索中,发现乙腈较甲醇可以更好地提高质谱响应,所以,选用乙腈作为有机相。水相则选择在0.02 mol/L醋酸铵溶液中加入0.1%甲酸,增加离子化效率,进一步改善峰形,使各组分完全分离。此外,采用电喷雾电离源(ESI)电离方式,发现替考拉宁在正离子模式下具有较高的离子化效率,且重现性良好。药物游离浓度的测定具有一定的难度,难点在于如何快速且有效地分离游离型药物,对血浆样品的处理有较高的要求。目前,测定药物游离浓度常见的样品处理方法主要有平衡透析法、超滤离心法[14]。其中最常用的是超滤离心法,该法不需稀释,不会改变生理pH值、离子组成,主要是利用离心力使游离药物通过半透膜,迅速而有效地从血液及其他生物样本中分离游离药物[15]。现国内外常用的超滤离心装置主要有Centrifree®和Amicon Ultra两种型号。本研究选用Centrifree®超滤装置进行分离。Roberts等在替考拉宁游离浓度测定的研究中,使用Amicon Ultra超滤装置分离蛋白[10]。但是,在实验前期探索中,我们发现Amicon Ultra装置所得血浆超滤液中加入乙腈后仍有沉淀析出,蛋白截留不完全;而Centrifree®装置产品说明书提示Centrifree®装置可以截留99.9%的蛋白,且使用该装置所得血浆超滤液中加入等量乙腈后未见明显沉淀析出。虽然两者所用滤膜均是再生纤维素膜,但是相比Amicon Ultra的凹槽设计、Centrifree®系列的平面设计,优点在于滤面更大,死体积更小,更有利于微溶质的滤过,是专用于从血液等生物样本中分离蛋白结合微溶质的,用于游离型药物的分离更为合理[15]。其次,为保证游离药物可以和蛋白有效分离,应选择合适的滤膜孔径。由于替考拉宁主要与白蛋白结合,而白蛋白的分子量约为66 000,本研究选用截留分子量为30 000的滤膜用以截留与白蛋白结合的药物,获得游离型替考拉宁。此外,确定合适的样品处理条件也是至关重要的。首先,收集的血浆样品为保持稳定性,需要低温保存,但温度较低可能会影响药物与蛋白的结合,导致药物游离浓度偏低,为保证测定结果的准确性,样品需在37 ℃平衡30 min后再进行超滤离心[10]。其次,离心力与离心时间也是影响超滤效果的重要因素[16]。离心力(1 000~2 000)×g可以达到最佳滤过率,离心时间在15~30 min时超滤液量与离心时间成正比[16],再结合文献[10]报道,本研究最终选用离心力1 500×g,离心时间30 min。同时,为保证样品测定尽可能不受基质的影响,本研究所有样品均采用空白血浆超滤液作为基质进行配制。有文献[17]也指出,游离药物分析最适宜的基质为空白血浆超滤液。方法建立后按照《中国药典》(2015年版)附录《生物样品定量分析方法验证指导原则》进行验证,验证结果均符合该指导原则的要求。综上,本研究建立了离心超滤结合UPLC-MS/MS检测替考拉宁游离浓度的方法,该方法简便、快速、灵敏、准确,可以为临床开展替考拉宁游离浓度监测提供依据。
-
表 1 桂皮醛加样回收率试验测定结果
称样量(g) 相当桂皮醛量(mg) 加入量(mg) 实测量(mg) 回收率(%) 平均回收率(%) RSD(%) 0.5078 0.1737 0.2038 0.3717 97.15 0.5045 0.1725 0.2038 0.3708 97.30 0.5064 0.1732 0.2038 0.3644 93.82 95.69 1.78 0.5018 0.1716 0.2038 0.3637 94.26 0.5051 0.1727 0.2038 0.3650 94.36 0.5006 0.1712 0.2038 0.3694 97.25  下载: 导出CSV
下载: 导出CSV
-
[1] 国家药典委员会. 中华人民共和国药典一部[S]. 北京: 化学工业出版社, 2020: 42.279. [2] 郑荣先. 通脉止渴胶囊薄层色谱鉴别[J]. 中国药物经济学, 2019, 14(6):47-50. doi: 10.12010/j.issn.1673-5846.2019.06.009 [3] 孙嘉惠, 张文晋, 赵振宇, 等. 植物分类学于中药鉴定学的意义[J]. 中药材, 2020, 43(9):2319-2323. [4] 陈海兰, 李晓华, 初洪波, 等. 穿山龙配方颗粒质量标准研究[J]. 中国现代中药, 2015, 17(7):712-715. [5] 刁兴彬, 高文学, 张芳, 等. 穿山龙提取物质量标准研究[J]. 中国药事, 2014, 28(8):871-874. [6] 米振清. 桂龙咳喘宁胶囊HPLC指纹图谱研究及7个活性成分的测定[J]. 中国药师, 2020, 23(5):950-954. doi: 10.3969/j.issn.1008-049X.2020.05.040 [7] 刘丹, 于丽红, 王颖, 等. 高效液相色谱同时测定桂生汤中毛蕊花糖苷和桂皮醛的含量[J]. 中南药学, 2019, 17(11):1903-1905. [8] 李耀华, 魏江存, 梁建丽, 等. 不同产地肉桂叶中香豆素、肉桂酸、桂皮醛成分的含量测定[J]. 中华中医药学刊, 2020, 38(2):54-57. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3177
- HTML全文浏览量: 1234
- PDF下载量: 16
- 被引次数: 0
