

 下载:
下载:
 下载:
下载:

-
光热疗法(photothermal therapy,PTT)是近年来新兴的肿瘤治疗技术,相比传统的手术治疗、化疗和放疗,具有更小的副作用[1-4]。PTT包含2个要素,即近红外光和光敏剂。近红外光具有较强的组织穿透能力[5-6],照射后肿瘤组织中的光敏剂可将光能转换为热能,当温度达到42 ℃以上时,可导致肿瘤细胞的凋亡或坏死[7]。
硫化铜(CuS)纳米粒是一种优良的光敏剂。相比广泛研究的金纳米粒,CuS纳米粒具有制备成本低的优点,特别是其具有稳定的光热效应。CuS纳米粒的近红外吸收主要源于Cu2+的d-d能级跃迁,因而其光热效应不受粒径、形状和生理环境的影响,可在肿瘤组织中维持稳定的光热效应[8]。CuS纳米粒具有良好的生物相容性,细胞毒性较低,但与其他多数无机或金属纳米粒同样存在体内发生蓄积的风险,长期使用的安全性未知。肾脏是处理纳米粒的主要器官之一,报道显示,直径小于6 nm的纳米粒可被肾脏滤过,从而使大部分纳米粒经尿液排出体外[9-10]。
因此,本文拟制备粒径小于6 nm的CuS纳米粒,通过深入分析各因素,获得最优的处方工艺,进一步通过光热曲线和细胞实验进行体外评价。
-
Litesizer500纳米粒度及Zeta电位分析仪(奥地利Anton Paar公司);JEOL 2010透射电子显微镜(日本电子株式会社);MDL-H-980-5W上转换发光用激光器(长春新产业光电技术有限公司);RX-300红外热成像仪(广东省东莞市不凡电子有限公司);NVMT SPARTAN夜视仪(梅越电子商务有限公司);DV215CD精密分析天平(美国Ohaus Discovery公司);HWS 26电热恒温水浴锅(上海一恒科学仪器有限公司);DF-101S集热式恒温加热磁力搅拌器(巩义市予华仪器有限责任公司);H1850R台式高速冷冻离心机(湖南湘仪实验室仪器开发有限公司);明澈-D24UV纯水/超纯水一体化系统(德国Merck Millipore公司)
-
二水合氯化铜(CuCl2·2H2O)、九水合硫化钠(Na2S·9H2O)(上海阿拉丁生化科技股份有限公司);聚维酮(polyvinylpyrrolidone,PVP,MW=10 000,默克生命科学有限公司);细胞增殖及毒性检测试剂盒(CCK-8,北京索莱宝生物技术有限公司);RPMI1640培养液,DMEM不完全高糖培养液,磷酸缓冲盐溶液(phosphate buffer saline,PBS)(江苏凯基生物技术有限公司);透析袋(MWCO 14000,美国Viskase公司);去离子水(实验室自制)。
-
参考文献确定CuS纳米粒基本处方[11]:取100 μl CuCl2溶液(300 mmol/L)加入10 ml PVP溶液(100 mg/ml)中,再加入30 μl Na2S溶液(1 mol/L),室温磁力搅拌5 min后,转入90 ℃水浴继续磁力搅拌15 min,测定其粒径和多分散指数(polydispersity index,PDI)。
-
考察搅拌时间、水浴温度、水浴时间、PVP浓度、CuCl2与Na2S摩尔比(Cu:S)对CuS纳米粒粒径和PDI的影响。
表1结果显示,搅拌时间越长,水浴温度越低,Cu:S越小,则CuS纳米粒粒径呈减小趋势。水浴时间越长,PVP浓度越大,粒径显示了先增大后减小的趋势。综合各因素不同水平对应的纳米粒粒径,发现水浴温度、PVP浓度和Cu:S易产生较大的粒径波动,表明其可能是影响CuS纳米粒粒径的主要因素。另外,各因素不同水平对PDI的影响均不显著。因此,根据单因素考察结果,确定搅拌时间为5 min,水浴时间为15 min,以粒径为评价指标,采用星点设计-响应面法进一步优化水浴温度、PVP浓度、Cu:S三个因素。
表 1 CuS纳米粒单因素考察结果(n=3)
因子 水平 粒子大小 (nm) 多分散指数 搅拌时间 (min) 3 18.13±5.42 0.27±0.01 5 16.12±1.29 0.27±0.01 7 15.40±1.86 0.26±0.01 水浴温度 (℃) 80 14.61±4.35 0.27±0.01 90 16.12±1.29 0.27±0.01 100 22.85±1.71 0.26±0.01 水浴时间 (min) 10 18.12±6.31 0.25±0.02 15 16.12±1.29 0.27±0.01 20 21.30±5.71 0.24±0.01 PVP浓度 (mg/ml) 80 25.56±4.40 0.27±0.01 100 16.12±1.29 0.27±0.01 120 20.72±3.38 0.25±0.02 Cu:S 3:1 15.08±2.92 0.26±0.01 2:1 20.25±1.16 0.27±0.01 1:1 16.12±1.29 0.27±0.01 1:2 11.34±3.08 0.25±0.02 1:3 11.27±2.88 0.25±0.02 -
在单因素考察的基础上,采用中心复合响应面设计(central composite design, CCD),选择上述筛选出的三个因素:水浴温度(X1)、PVP浓度(X2)、Cu:S(X3)为考察因素,粒径(Y1)为评价指标,进行三因素五水平的星点设计。
-
星点实验设计的因素、水平及代码值如表2所示,实验设计与结果如表3所示。
表 2 星点设计因素和水平
因子 水平 −1.682 −1 0 +1 +1.682 X1 (℃) 60.00 70 85 100 110.20 X2 (mg/ml) 66.36 80 100 120 133.64 X3 0.25 0.33 1.67 3 4 表 3 星点设计实验安排表和结果(n=3)
序号 X1 (℃) X2 (mg/ml) X3 Y1 (nm) 1 −1 −1 −1 13.04 2 +1 −1 −1 14.23 3 −1 +1 −1 14.48 4 +1 +1 −1 13.39 5 −1 −1 +1 24.37 6 +1 −1 +1 27.30 7 −1 +1 +1 18.62 8 +1 +1 +1 20.73 9 −1.682 0 0 18.87 10 +1.682 0 0 15.62 11 0 −1.682 0 24.01 12 0 +1.682 0 17.06 13 0 0 −1.682 13.92 14 0 0 +1.682 15.11 15 0 0 0 18.81 16 0 0 0 16.84 17 0 0 0 16.14 18 0 0 0 17.89 19 0 0 0 16.20 20 0 0 0 16.30 -
采用Design expert 8.0.6,将各影响因素(X1、X2、X3)和评价指标(Y1)进行二次多项式模型拟合,方程如下:
Y1=4.15−0.0086X1−0.18X2+0.42X3−0.045X1X2+0.064X1X3−0.17X2X3+0.043X12+0.17X22−0.22X32
此时二项式模型达显著水平(P=0.006 5),r=0.9129,说明该方程与实际情况拟合度良好。
-
根据表3结果,绘制水浴温度(X1)、PVP浓度(X2)及Cu:S(X3)对粒径(Y1)的响应面。
根据图1的数据进行分析,确定最优参数为:水浴温度100 ℃,PVP浓度为100 mg/ml,Cu∶S为1∶3。按照最优处方工艺平行制备3份样品,粒径分别为9.37、12.39、9.84 nm,平均值为(10.53±1.63)nm,预测值为10.86 nm,根据公式:偏差=(预测值−实际值)/预测值×100%,得出实际值与预测值的偏差为3.04%,说明建立的数学模型较为可靠,预测性良好。
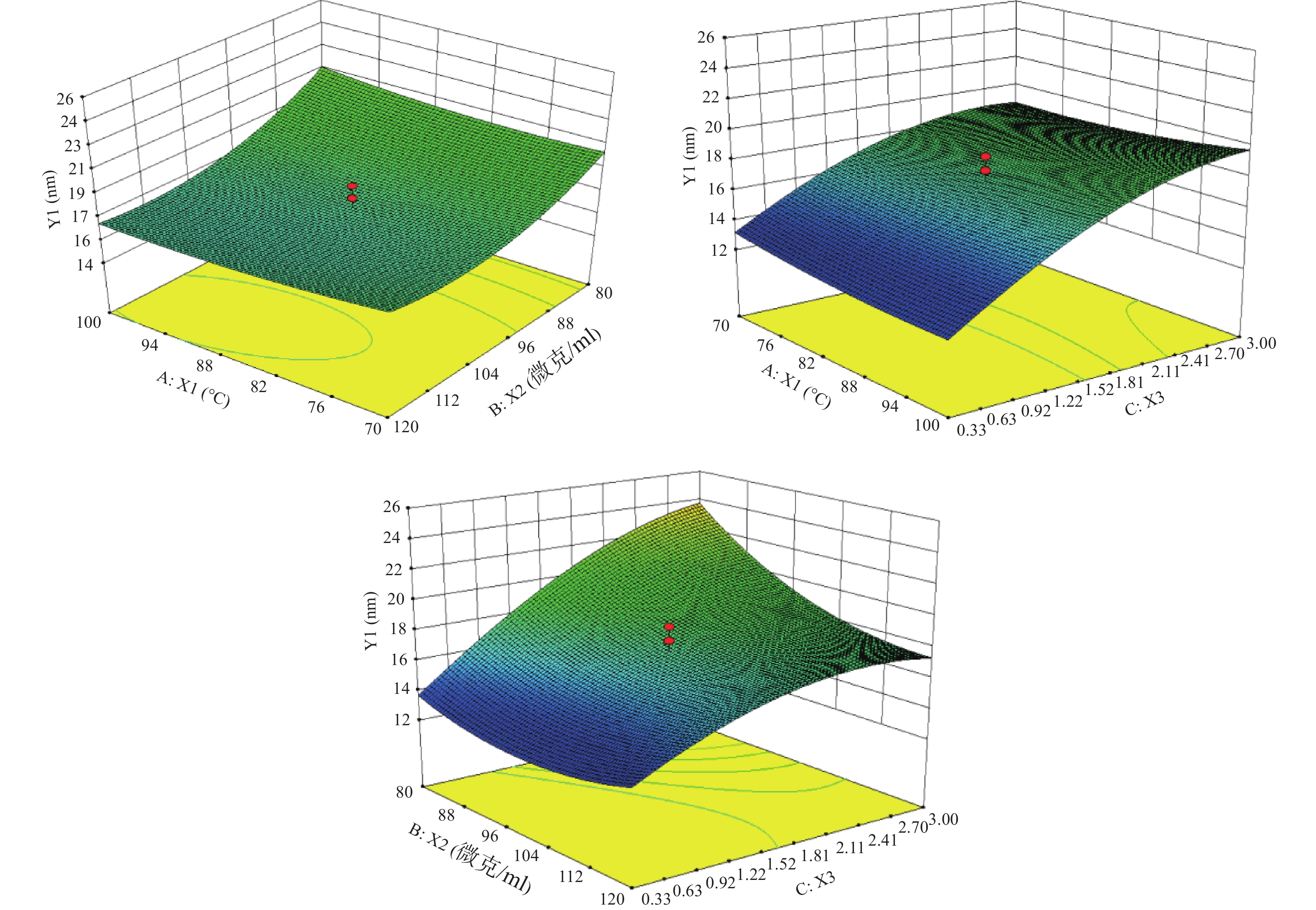
图 1 温度(X1)、PVP浓度(X2)、Cu:S(X3)对粒径值(Y1)的三维响应面图
-
取CuS纳米粒适量,加去离子水稀释10倍,取5 μl稀释后的CuS纳米粒,滴于缚有碳支持膜的铜网上,铜网下铺滤纸,用于吸去多余的液体。自然晾干后,用透射电子显微镜(transmission electron microscope, TEM)观察纳米粒形态。图2结果显示,CuS纳米粒形态圆整,分散性良好,经Image J软件测量,平均粒径为(3.10±0.81)nm。

图 2 CuS纳米粒TEM图
-
将CuS纳米粒分别放置在4 ℃与25 ℃条件下,定时测定粒径。经统计发现,20 d内CuS纳米粒的粒径变化没有明显差异,说明所制的CuS纳米粒具有良好的粒径稳定性(图3)。
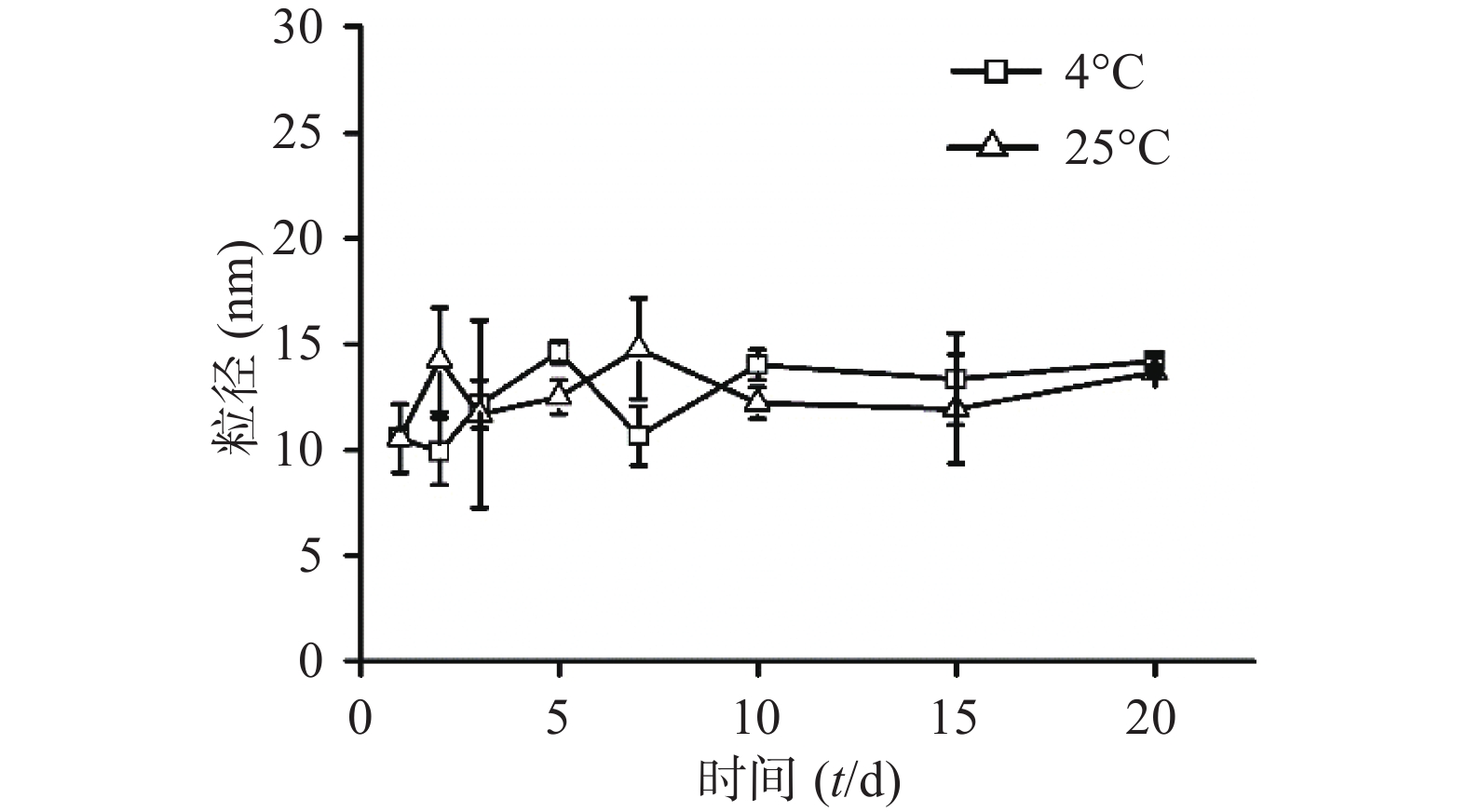
图 3 CuS纳米粒分别在4 ℃与25 ℃下的粒径变化曲线(n=3)
-
⑴近红外光功率密度:考察CuS纳米粒浓度为50 μg /ml条件下,不同近红外光功率密度(0.5 、1、1.5、2 W/cm2)对CuS纳米粒光热效应的影响。采用980 nm的近红外激光器,红外热成像仪每隔30 s记录温度,记录10 min,平行测量3组,绘制升温曲线,同时,以水在相同条件下做对照。随着功率密度增大,CuS纳米粒升温加快(图4),而在相同条件下,去离子水的升温速率显著低于CuS纳米粒(图5)。
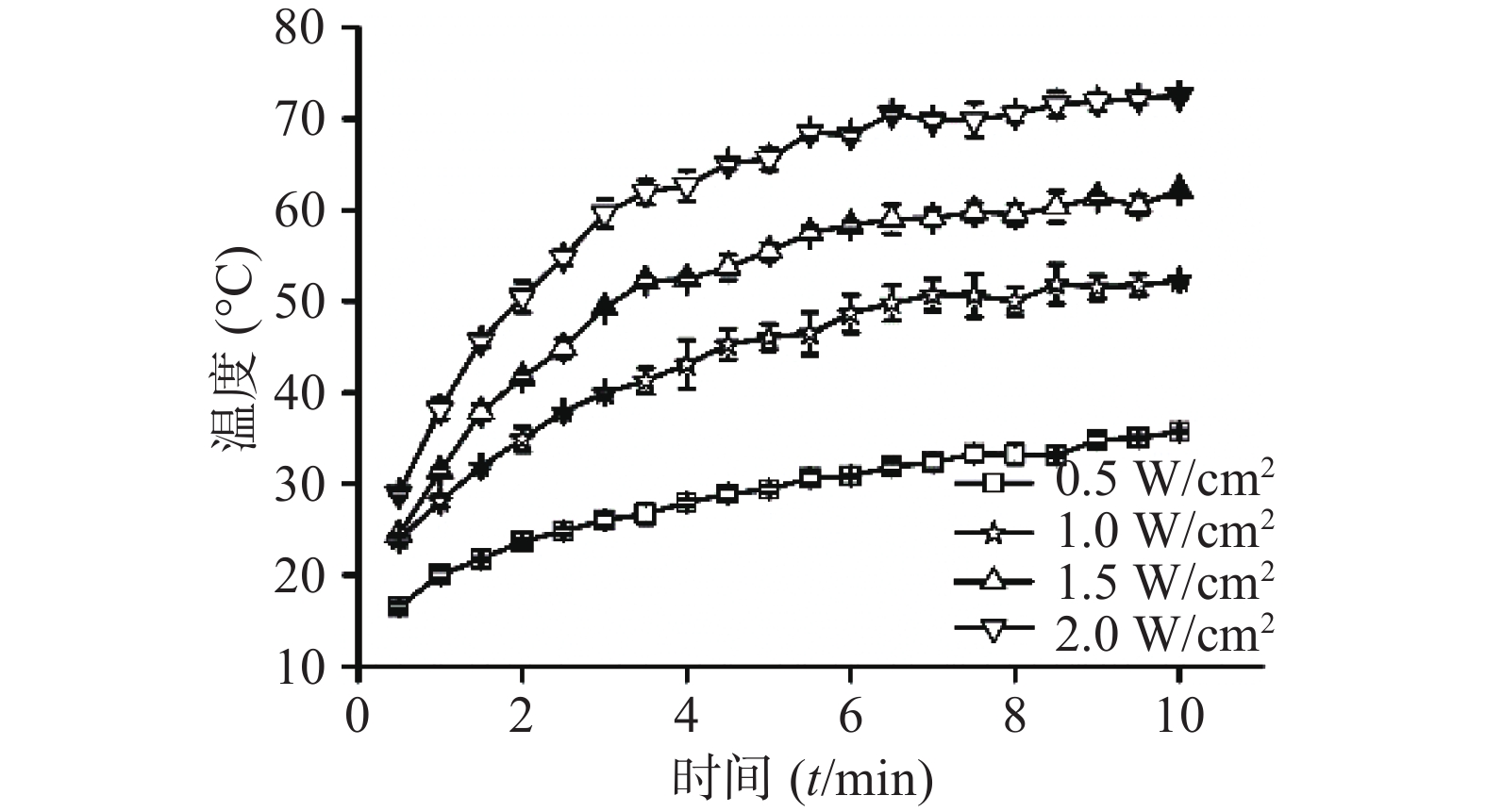
图 4 CuS纳米粒在不同功率密度下的升温曲线(n=3)
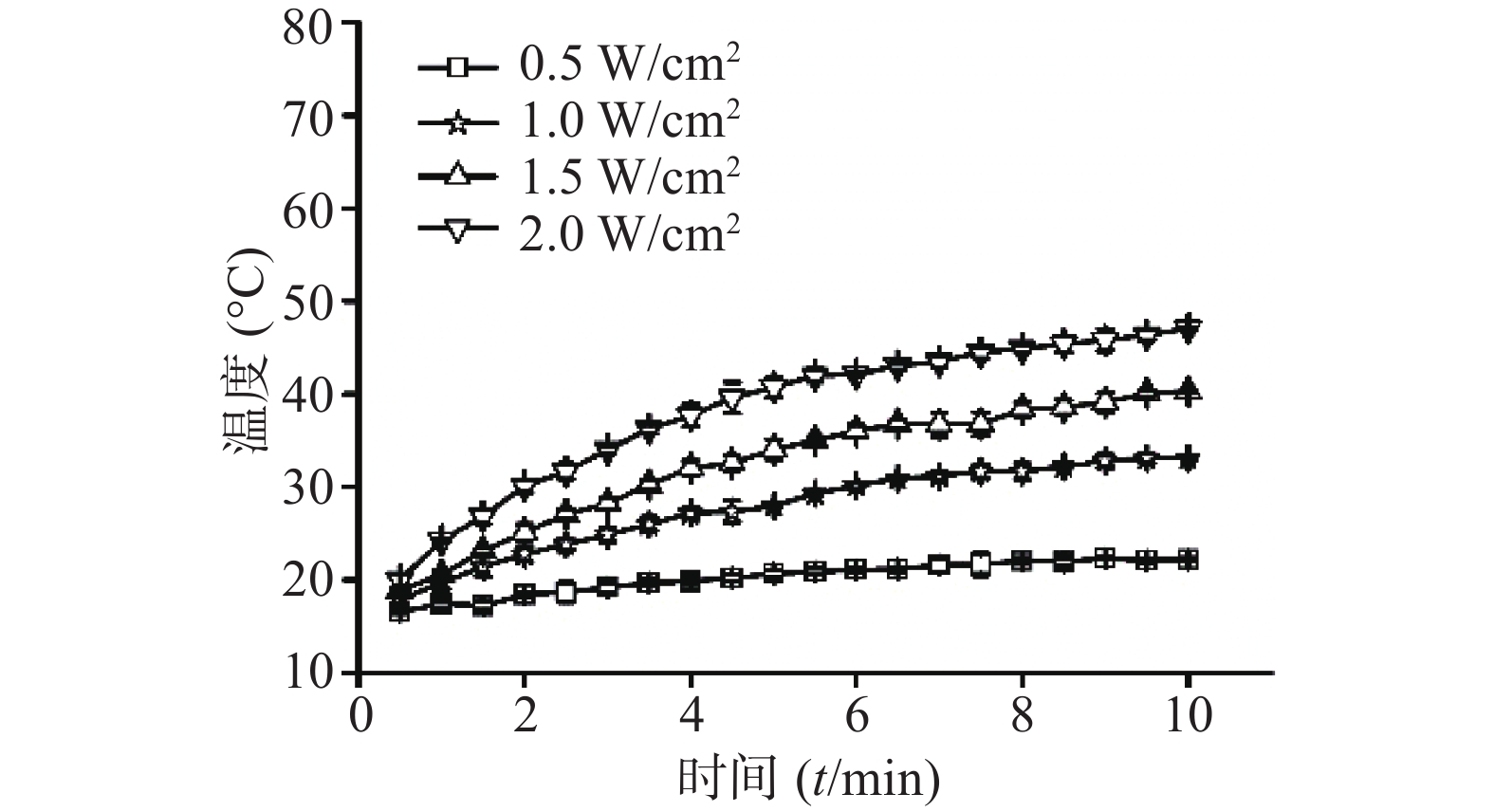
图 5 水在不同功率密度下的升温曲线(n=3)
⑵CuS纳米粒浓度:考察在1 W/cm2功率密度下,CuS纳米粒不同浓度(25 、50 、150 、200 μg/ml)对其光热效应的影响,并以去离子水为空白对照。采用980 nm的近红外激光器,红外热成像仪每隔30 s记录温度,记录10 min,平行3组,绘制升温曲线。结果显示,在相同功率密度条件下,CuS纳米粒浓度越大,升温速率越大(图6)。
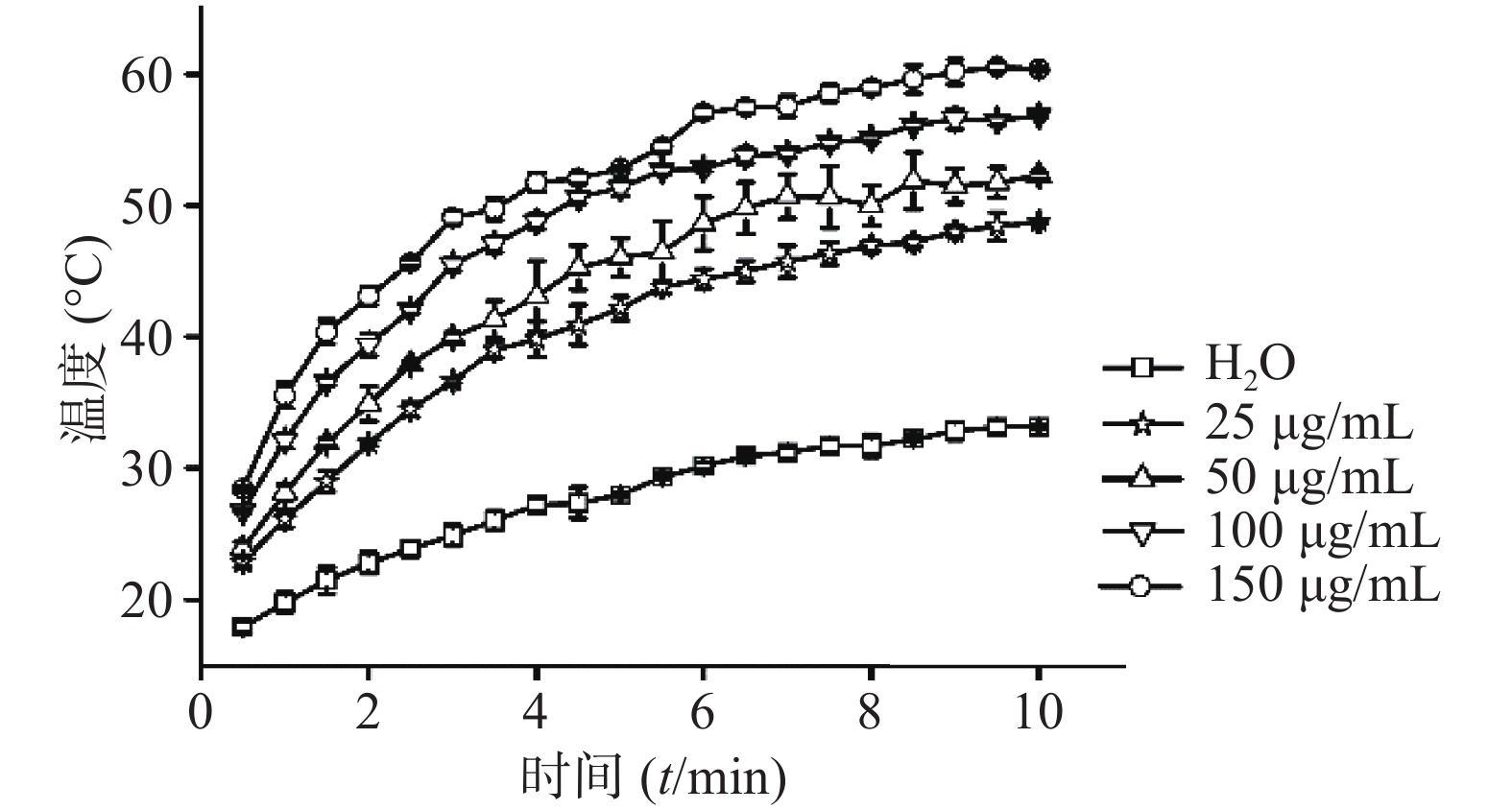
图 6 CuS纳米粒在不同浓度下的升温曲线(n=3)
-
采用980 nm近红外激光器,选取CuS浓度为50 μg /ml,激光功率密度为1 W/cm2,将激光器“开启-关闭”4个循环,即照射10 min后停止,温度恢复至室温后再次开启照射。如图7所示,CuS纳米粒经近红外光照射4个循环的温度变化曲线基本一致,即反复照射不影响CuS纳米粒的光热效应,表明其具有良好的光热稳定性。
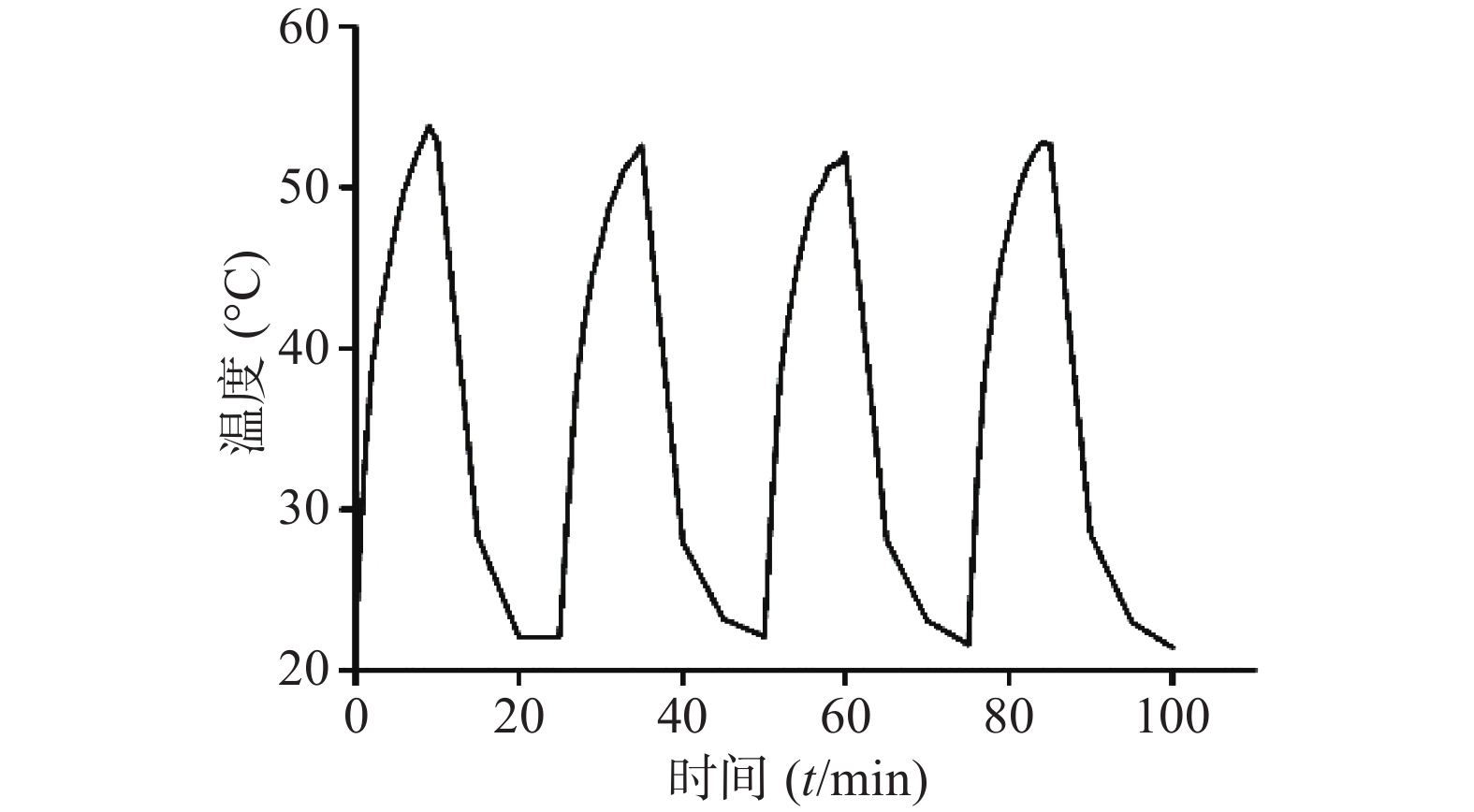
图 7 CuS纳米粒经近红外光照射4个周期的温度曲线
-
考察CuS纳米粒对正常人肾小管上皮细胞(HK2)以及乳腺癌细胞(4T1)的毒性。实验前,取适量CuS纳米粒放入透析袋(MWCO=14000)中,透析24 h以除去杂质。取对数生长期细胞,调整HK2细胞浓度为1×105 cells/ml,4T1细胞浓度为5×104 cells/ml,将100 μl/孔的细胞悬液接种于96孔细胞培养板,置于37 ℃,5% CO2培养箱中。孵育24 h后,每孔加入10 μl不同浓度的CuS纳米粒,使终浓度分别为3.1、6.3、12.5、25、50、100、150及200 μg/ml,继续培养24 h后,去除96孔板中培养基,加PBS洗涤2次(以排除Cu2+对CCK-8试剂的影响),每孔加入100 μl培养基及10 μl CCK-8试剂,于培养箱中孵育4 h后,用酶标仪测定450 nm处的吸光度值(A)。参照公式:
细胞活力(%)=(A加药−A空白)/(A0加药−A空白)×100%,
计算细胞活力百分率,其中A加药指具有细胞、CCK-8溶液和药物溶液的孔的吸光度,A空白指具有培养基和CCK-8溶液而没有细胞的孔的吸光度,A0加药指具有细胞、CCK-8溶液而没有药物溶液的孔的吸光度。
结果显示(图8),CuS纳米粒分别在100 μg/ml及150 μg/ml浓度范围内,对4T1乳腺癌细胞以及HK2肾细胞均无显著的细胞毒性(细胞活力大于80%),表明CuS纳米粒具备良好的生物相容性。
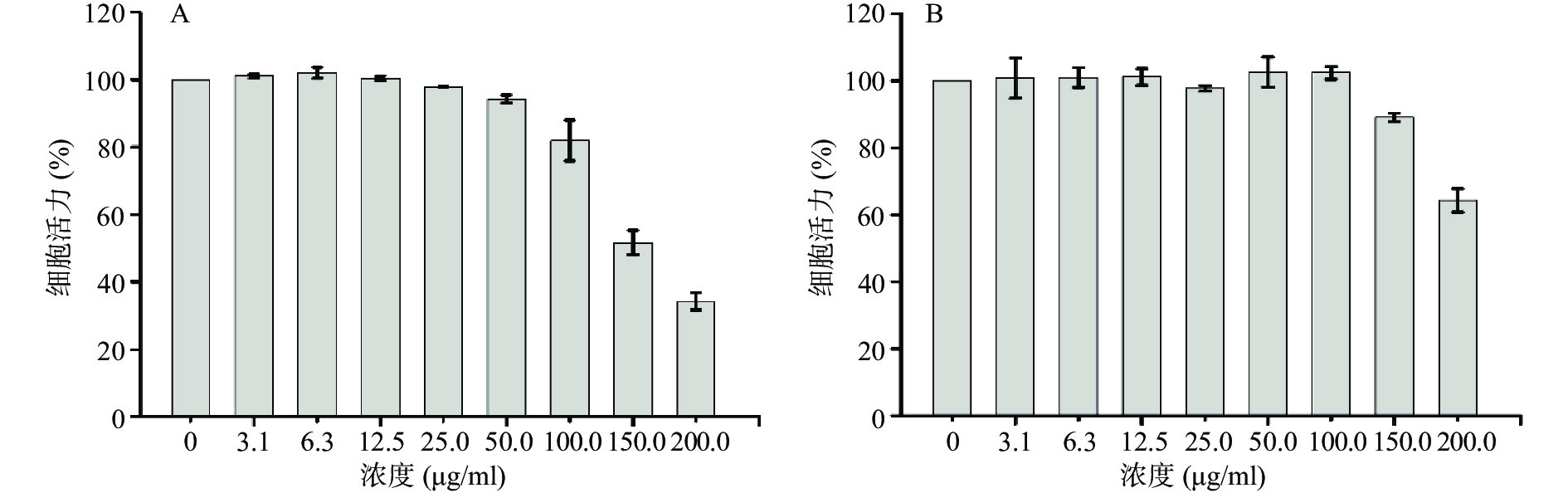
图 8 CuS纳米粒浓度对细胞活力影响
-
考察CuS纳米粒的光热效应对4T1细胞的杀伤作用。取对数生长期细胞,调整4T1细胞浓度为5×104 cells/ml,将200 μl/孔细胞悬液接种于96孔细胞培养板,置于37 ℃,5% CO2培养箱中培养24 h后,去除全部培养基,加入200 μl含CuS纳米粒的培养基(CuS纳米粒的浓度为50 μg /ml)。采用1 W/cm2的近红外光,照射不同时间(0、1、3、5、7、10 min),随后采用CCK-8试剂测定细胞活力百分率,参照“2.5.1”项下公式计算。结果显示(图9),近红外光照射1 min对4T1细胞的活力无明显影响,继续延长照射时间,细胞活力呈显著的降低趋势,照射10 min后的细胞活力仅为28.76 %。
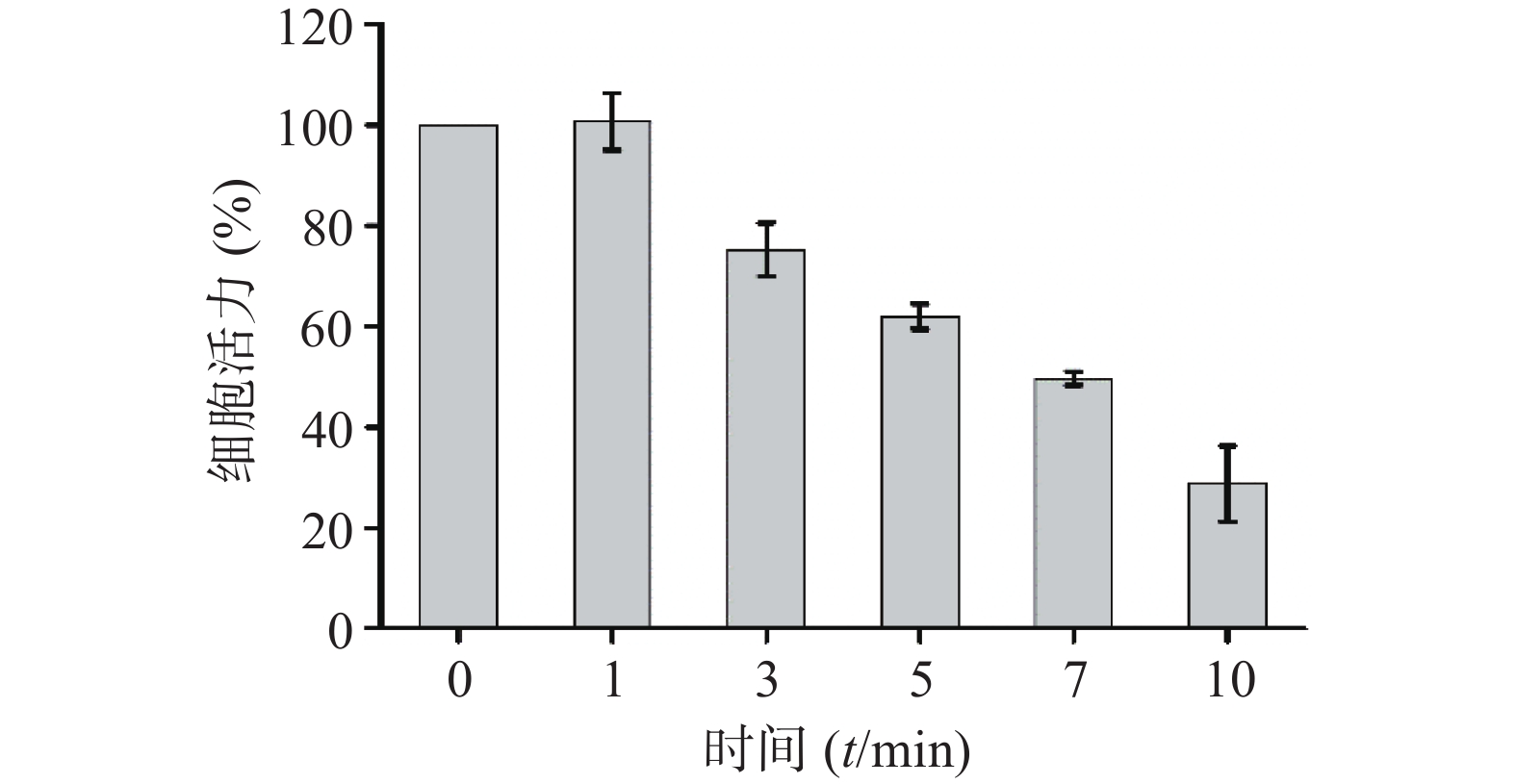
图 9 CuS纳米粒光热效应作用下的4T1乳腺癌细胞的存活率(n=5)
采用与上述相同的方法,改为6孔板,经980 nm近红外光(1 W/cm2)照射10 min后,加入0.2 %台盼蓝对死细胞进行染色(2 min)。去除染液并用PBS清洗2遍后,在倒置相差显微镜下进行观察。如图10所示,激光照射区域,细胞被染为蓝绿色,且显示了明显的边界,而对照组不加CuS纳米粒,仅给予近红外光照射,只有个别零散的细胞着色。图9与图10结果均表明CuS纳米粒对肿瘤细胞具有显著的光热杀伤效果。
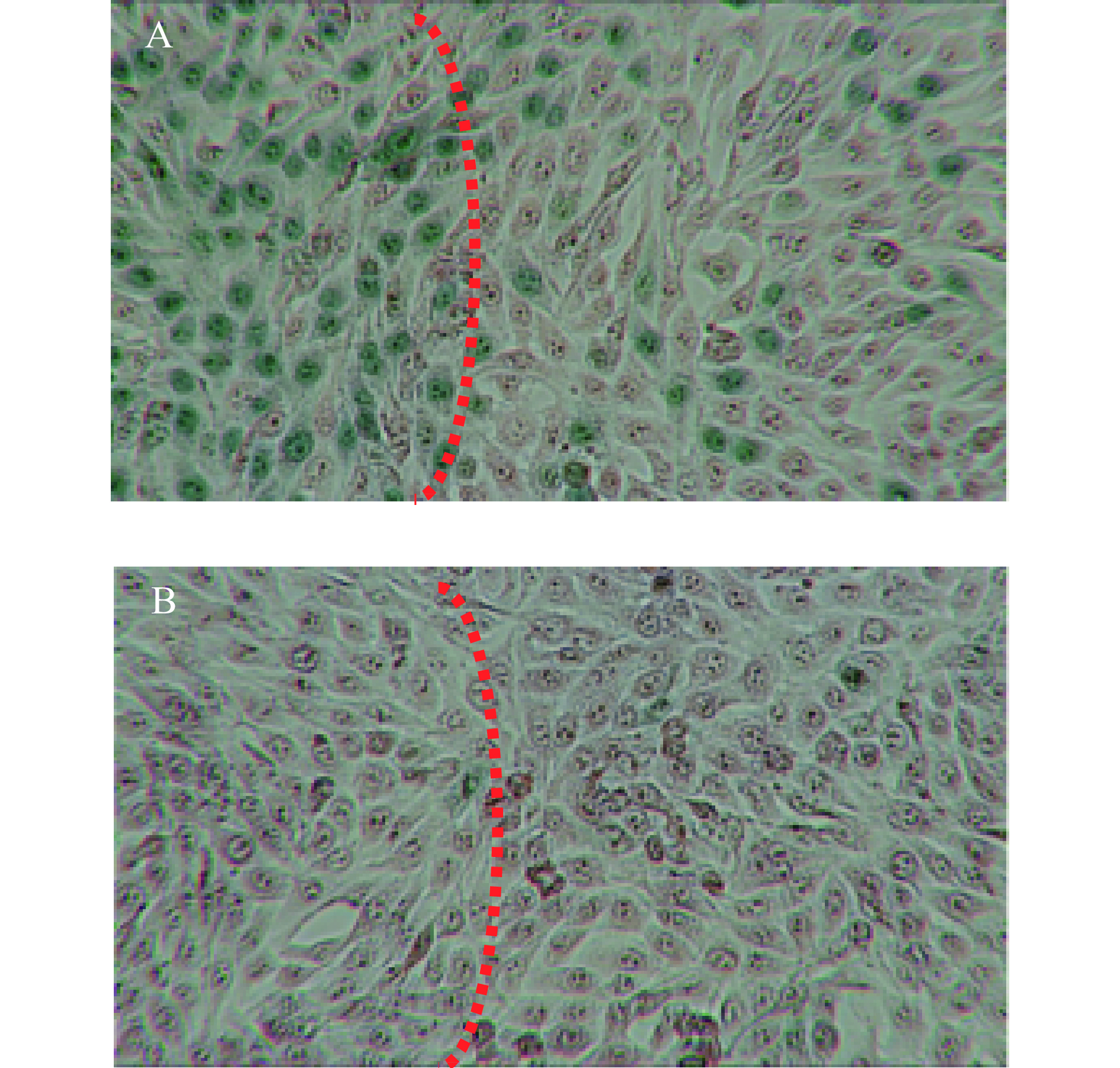
图 10 4T1乳腺癌细胞光热杀伤台盼蓝染色图片
-
目前光热治疗作为新型的肿瘤治疗技术,寻找合适的光敏剂是亟待解决的重要问题。光热稳定、价格低廉等是CuS纳米粒的主要优点,但目前文献所报道的CuS纳米粒粒径多在10 nm以上[12-13],可能引起CuS纳米粒在体内的蓄积,将限制其在临床上的使用,而减小粒径可以有效解决这个问题。本研究经单因素考察和星点设计-响应面优化法,得到CuS纳米粒的最优处方工艺,成功制备实际粒径为(3.10±0.81)nm的CuS纳米粒,且制备方法简单,易于工业化生产。该处方中以PVP作为保护剂,使CuS纳米粒具有良好的粒径稳定性,分散度高,不易发生聚集沉淀,单因素考察分析也可发现PVP浓度对CuS纳米粒粒径具有较大影响,PVP浓度低,CuS纳米粒容易发生聚集,粒径增大;浓度太高,保护层过厚,也会引起CuS纳米粒粒径增大,因此选择合适的PVP浓度,是控制CuS纳米粒粒径关键因素之一。
由于CuS纳米粒在近红外区域(700~1 400)nm具有强吸收,使其在近红外光照射下,具有较强的光热效应,所优选的CuS纳米粒在1 W/cm2的激光功率密度下,照射4 min,温度即可达到42 ℃以上,可引起肿瘤细胞的凋亡或坏死[14]。因CuS纳米粒的光热转换性能来源于Cu2+的d-d能级跃迁,不易受外界环境的影响,所优选的CuS纳米粒具有良好的光热稳定性。本实验所优选的CuS纳米粒生物相容性好,且对肿瘤细胞具有显著的光热杀伤作用,这与多数文献报道相一致[15-16]。
综上,本研究所制备的CuS纳米粒粒径有望解决CuS纳米粒体内蓄积的问题,使其更好地应用于肿瘤光热治疗。本课题将进一步研究所制硫化铜纳米粒在体内的代谢情况以及对肿瘤组织的光热杀伤效果。
Optimization of formulation process and in vitro evaluation of copper sulfide nanoparticles
-
摘要:
目的 为避免硫化铜(CuS)纳米粒体内蓄积,制备并优化CuS纳米粒,分析粒径影响因素并评价其光热性能。 方法 在单因素考察基础上,采用星点设计-响应面法进一步分析各因素对CuS纳米粒粒径的影响,获得最佳处方工艺。考察优选的纳米粒的理化性质,包括形态、粒径稳定性、光热转换性能及光热稳定性。采用CCK-8法评价CuS纳米粒对4T1乳腺癌细胞以及HK2肾细胞的毒性,并考察其光热效应对4T1乳腺癌细胞的杀伤效果。 结果 CuS纳米粒优选处方工艺的水合粒径为(10.53±1.63)nm;透射电镜下显示其粒径为(3.10±0.81)nm;所优选的CuS纳米粒具有良好的粒径稳定性,良好的光热转换性能与光热稳定性。细胞毒研究显示,所优选的CuS纳米粒分别在100 μg/ml及150 μg/ml浓度范围内,对4T1乳腺癌细胞以及HK2肾细胞均无显著的细胞毒性,且其对4T1乳腺癌细胞具有显著的光热杀伤效果。 结论 所制备的CuS纳米粒实际粒径小于6 nm且具有良好的光热效应,有望解决CuS纳米粒体内蓄积的问题,使其更好地应用于抗肿瘤治疗。 Abstract:Objective To avoid the accumulation of copper sulfide (CuS) nanoparticles, prepare and optimize CuS nanoparticles, analyze the factors affecting the particle size and evaluate their photothermal properties. Methods Based on the single factor study, central composite design-response surface methodology was used to optimize the CuS nanoparticle formulation process. The morphology, particle size stability, photothermal conversion efficiency, photothermal stability of optimized CuS nanoparticles were characterized. The toxicity of CuS nanoparticles on 4T1 breast cancer cells and HK2 kidney cells was evaluated by CCK-8 method. In vitro photothermal experiment was used to investigate the ability of CuS nanoparticles on killing 4T1 breast cancer cells. Results The average hydration dynamic diameter of optimized CuS nanoparticles was (10.53±1.63)nm, the actual particle size of CuS nanoparticles showed by TEM image was (3.10±0.81)nm. It had good particle size stability, good photothermal conversion efficiency and photothermal stability. Within the concentration range of 100 μg/ml and 150 μg/ml, it showed no significant toxicity on 4T1 breast cancer cells and HK2 kidney cells, indicating the good stability of CuS nanoparticles. In vitro photothermal therapy showed that CuS nanoparticles had good ability to kill 4T1 breast cancer cells by photothermal. Conclusion The prepared CuS nanoparticles have a small particle size (less than 6nm) and a good photothermal effect, which is expected to solve the problem of CuS nanoparticles accumulation in vivo and make it better for tumor treatment. -
结直肠癌(CC)发生率占所有肿瘤发生率的第3位,病死率仅次于肺癌,属于下消化系统恶性肿瘤[1]。目前,结肠癌的治疗主要采用多学科的综合治疗模式[2]。包括5-氟尿嘧啶、奥沙利铂与贝伐珠单抗等的组合[3-5]。然而,这些常规的化疗方案可能造成患者不耐受以及骨髓等的抑制。因此,寻找新的有效的药物对于现今结肠癌治疗具有重要意义。
紫杉醇(PTX)是一种广谱抗肿瘤活性的化疗药物,来源于太平洋紫杉树皮(红豆杉)[6,7]。临床试验结果表明,PTX在几种癌症治疗中有较好的活性,包括:乳腺癌、皮肤恶性肿瘤、非小细胞肺癌以及卵巢癌等[8,9]。然而,由于结肠癌中过表达的P糖蛋白(P-gp)引起了多重耐药性(MDR),导致PTX对结肠癌临床治疗效果不甚理想[10]。此外,PTX生物半衰期短,其一代药物Taxol以聚氧乙烯蓖麻油为表面活性剂,具有引起患者过敏反应的风险,从而限制了PTX临床疗效的发挥[11]。因此,迫切需要对PTX进行结构改进与剂型设计,改善其药物递送效率,提高生物利用度[12]。
脂肪酸作为生物膜和生物信号分子的重要成分,参与了细胞能量产生、代谢的过程。由于肿瘤细胞增殖快速,需要大量的细胞合成物质和能量的供应,因此,患有恶性肿瘤的患者其脂肪酸合成也较快。其中,肿瘤细胞因为迅速繁殖对含有16个碳原子的棕榈酸(PA)为主的脂肪酸需求量大,因此利用PA进行修饰有利于药物被肿瘤细胞摄取[13,14]。再者,研究发现,由PA修饰的紫杉醇,能降低其对P-gp的亲和力,避免了PTX进入肿瘤细胞后的外排,提高紫杉醇对于结肠癌治疗的有效性[15]。
此外,作为一种成熟的药物载体,脂质体(Lip)在体内可被降解、无毒性和免疫原性,能够增强药物在体内的稳定性,从而可以减少给药剂量、降低毒副作用,并且其表面具有可修饰性[16],例如,采用聚乙二醇磷脂(PEG-DSPE)修饰的脂质体因其空间位阻效应可延长其在体内的循环时间[17]。同时结合肿瘤组织的增强通透性和滞留效应(EPR),使药物通过被动靶向递送到靶部位[18]。
结合以上背景,本研究首先通过棕榈酸酯与紫杉醇共价键结合构建紫杉醇棕榈酸酯(PTX-PA),并建立基于高效液相(HPLC)的定量分析方法,旨在降低其对P-gp的亲和力,避免PTX进入肿瘤细胞后被外排,从而改善紫杉醇毒性较大、生物半衰期短、成药性差等问题,提高紫杉醇对于结肠癌治疗的有效性。其次,我们将PTX-PA包载进PEG修饰的脂质体构建紫杉醇棕榈酸酯的脂质体(PTX-PA/Lip),以实现其长循环,增加PTX的疗效、降低其毒副作用。最后,采用工艺筛选与单因素处方优化的方法制备最佳PTX-PA/Lip,为PTX-PA的制剂学研究奠定基础[19]。
1. 仪器与材料
1.1 仪器
高效液相色谱仪(安捷伦科技有限公司,美国);十万分之一电子天平(MS105DU,梅特勒托利多公司,瑞士);超滤管(30 kD,密理博公司,美国);低温高速离心机(Eppendorf-200,艾本德公司,德国);高压均质机(NanoGenizer,美国);Zeta-sizer Nano粒度仪(Nano-ZS,马尔文公司,英国)。
1.2 试剂与材料
紫杉醇购自江苏红豆杉生物科技股份有限公司,纯度≥98%;棕榈酸购自中国医药集团上海化学试剂公司,纯度≥99%;蛋黄卵磷脂(PC98-T)、胆固醇、DSPE-PEG 2000均购自上海艾韦特医药科技有限公司;1-(3-二甲氨基丙基)-3-乙基碳二亚胺(EDC)、4-二甲氨基吡啶(DMAP)、乙酸乙酯、二氯甲烷、无水乙醇、石油醚均购自中国医药集团上海化学试剂公司,分析纯;乙酸乙酯、PC-98T蛋黄卵磷脂(上海艾韦特医药科技有限公司);胆固醇(上海艾韦特医药科技有限公司);二氯甲烷、DSPE-PEG 2000(上海艾韦特医药科技有限公司);无水乙醇、石油醚购自中国医药集团上海化学试剂公司;甲醇购自美国默克公司,色谱纯。
2. 方法
2.1 PTX-PA的制备与纯化
PTX-PA前药由PTX与PA发生酯化反应合成,其合成过程如下[20,21]:精密称取PTX 0.85 g,加入无水二氯甲烷 30 ml。依次加入精密称取的EDC 0.19 g、DMAP 0.15 g和PA 0.31 g,在氮气保护下室温搅拌反应12 h。反应结束后,用5%柠檬酸水溶液、饱和食盐水洗涤3遍,旋蒸除去有机溶剂,即得PTX-PA粗品。采用石油醚和乙酸乙酯通过柱层析法对PTX-PA进行分离、纯化。洗脱完成后,将产物旋蒸去除有机溶剂,得到的白色固体即为PTX-PA,样品经核磁共振氢谱、碳谱确定为PTX-PA,样品纯度98.5%。
2.2 基于HPLC定量检测的方法学建立[22]
2.2.1 最大吸收波长测定与色谱条件设定
精密取适量的PTX-PA粉末,采用甲醇溶解,定容,通过全波长(190~400 nm)扫描测定其紫外最大吸收波长λmax。
本实验采用Agilent Eclipse plus C18色谱柱(4.6 mm×250 mm,5 μm),流动相采用甲醇/水(95∶5,V/V),进样量20 μl,流速1.0 ml/min。
2.2.2 样品溶液配制
空白溶液的配制:精密量取不含药的空白脂质体1 ml加入适量的甲醇溶液,超声,用甲醇定容至25 ml,0.45 μm的微孔滤膜过滤,滤液即为空白溶液。
对照品溶液的配制:精密称取PTX-PA 粉末50 mg,用甲醇溶解并定容至50 ml,过滤,即得PTX-PA对照品储备溶液。
供试品溶液的配制:精密量取1 ml PTX-PA/Lip,加入适量甲醇溶解、超声破乳、定容至25 ml,过滤,滤液即为供试品溶液。
2.2.3 专属性考察
分别取适量空白溶液、对照品溶液和供试品溶液,用流动相稀释至适当的浓度后,按照“色谱条件”中建立的高相液相参数进行进样分析。
2.2.4 线性关系考察
精密量取适量PTX-PA对照品储备液依次稀释为浓度1、5、10、25、50、100 μg/ml的PTX-PA系列浓度。按照“色谱条件”中的高效液相参数进行进样分析(n=5),并记录不同浓度的PTX-PA的色谱峰面积。以浓度(C)为X轴、峰面积(A)为Y轴进行回归。
2.2.5 精密度考察
日内精密度考察:精密吸取3种不同浓度(5、25、100 μg/ml)的 PTX-PA对照品溶液,对同一浓度溶液连续进样5次,每次10 µl,按照“2.2.1”项下色谱条件进行分析,记录各个浓度吸收峰面积。
日间精密度考察:精密吸取3种不同浓度(5、25、100 μg/ml)的 PTX-PA对照品溶液,对同一浓度溶液连续进样5 d,每天1次,每次10 µl,按照“2.2.1”项下色谱条件进行分析,记录第0、1、2、3、4天的吸收峰面积,计算样品浓度,考察仪器的日间精密度。
2.2.6 重复性与稳定性
空白脂质体超声破乳,配制成3种不同浓度(1、10、100 μg/ml),连续进样5次,每次10 µl,记录各吸收峰面积,考察重复性。精密吸取浓度为50 μg/ml的 PTX-PA供试品溶液,于0、2、4、6、8、12、24 h进样测定,每次进样10 µl,记录各紫外吸收峰面积,考察样品的稳定性。
2.2.7 加样回收率考察
空白脂质体超声破乳,分别加入浓度为1 mg/ml的PTX-PA溶液0.5、2.5、5 ml,采用流动相稀释定容至100 ml,分别进样,并结合PTX-PA的理论浓度(5、25、50 μg/ml)进行加样回收率的计算与分析。
2.3 紫杉醇棕榈酸酯脂质体PTX-PA/Lip的制备
2.3.1 PTX-PA/Lip的制备方法的选择
(1) 薄膜分散法
精密称取PTX-PA 20 mg、蛋黄卵磷脂350 mg、胆固醇5 mg和DSPE-PEG2000 25 mg,加入适量的二氯甲烷使其充分溶解。然后旋蒸除去有机溶剂,使圆底烧瓶底部形成一层均匀透明的薄膜,再加入预热至同等温度的重蒸水10 ml,震荡、水化,得PTX-PA/Lip粗品。将得到的粗品经探头超声(1 min)、过滤处理后,即得PTX-PA/Lip纳米给药系统[19]。
(2) 高压均质法
将薄膜分散法制得的PTX-PA/Lip粗品置于高压均质机中,经3次均质处理后(均质压力12 000 psi),即得PTX-PA/Lip纳米给药系统[23,24]。
(3)挤出法
将薄膜分散法制得的PTX-PA/Lip粗品置于挤出器中,使分别经过孔径为0.2、0.1、0.05 μm的聚碳酸酯膜,即得PTX-PA/Lip纳米给药系统[25-27]。
2.3.2 处方工艺筛选
(1)磷脂种类的选择
采用薄膜分散法考察不同磷脂制备的PTX-PA/Lip,包括:氢化磷脂(HSPC)、蛋黄卵磷脂(PC98-T)、蛋黄磷脂(EPCS)、二棕榈酸磷脂酰胆碱(DPPC),以形态、粒径、包封率为指标进行评价。
(2)磷脂和药物/胆固醇比例的考察
分别以磷脂和药物的质量比(5∶1、10∶1、20∶1、30∶1、40∶1)/PC98-T和胆固醇的质量比(4∶0.05、4∶0.1、4∶0.2、4∶0.3、4∶0.4、4∶0.5)为自变量制备PTX-PA/Lip脂质体,考察不同处方的粒径、粒径分布、包封率,确定处方中磷脂与药物/胆固醇的质量比。
(3)药物和DSPE-PEG2000比例的考察
以PTX-PA和DSPE-PEG2000的质量比(1∶0.5、1∶1、1∶1.5、1∶2、1∶2.5)为自变量,考察其用量对制剂的外观澄明度、纳米粒子大小等是否产生影响。
(4)薄膜蒸发法的温度考察
将旋转蒸发仪温度分别设置为35、40、45、50、55 ℃,考察薄膜蒸发过程中温度对PTX-PA/Lip形态、粒径、包封率等的影响。
(5)探头超声时间的考察
以探头超声PTX-PA/Lip粗品的时间为自变量,考察不同超声时间(30、60、90、180、240 s)对PTX-PA/Lip纳米制剂形态、纳米粒子大小的影响。
2.4 统计学分析
数据采用IBM SPSS Statistics 27.0进行统计分析,采用单因素方差分析ANOVA进行显著性检验与评价。
3. 结果与讨论
3.1 基于PTX-PA的HPLC定量检测方法学建立
3.1.1 最大吸收波长的选择
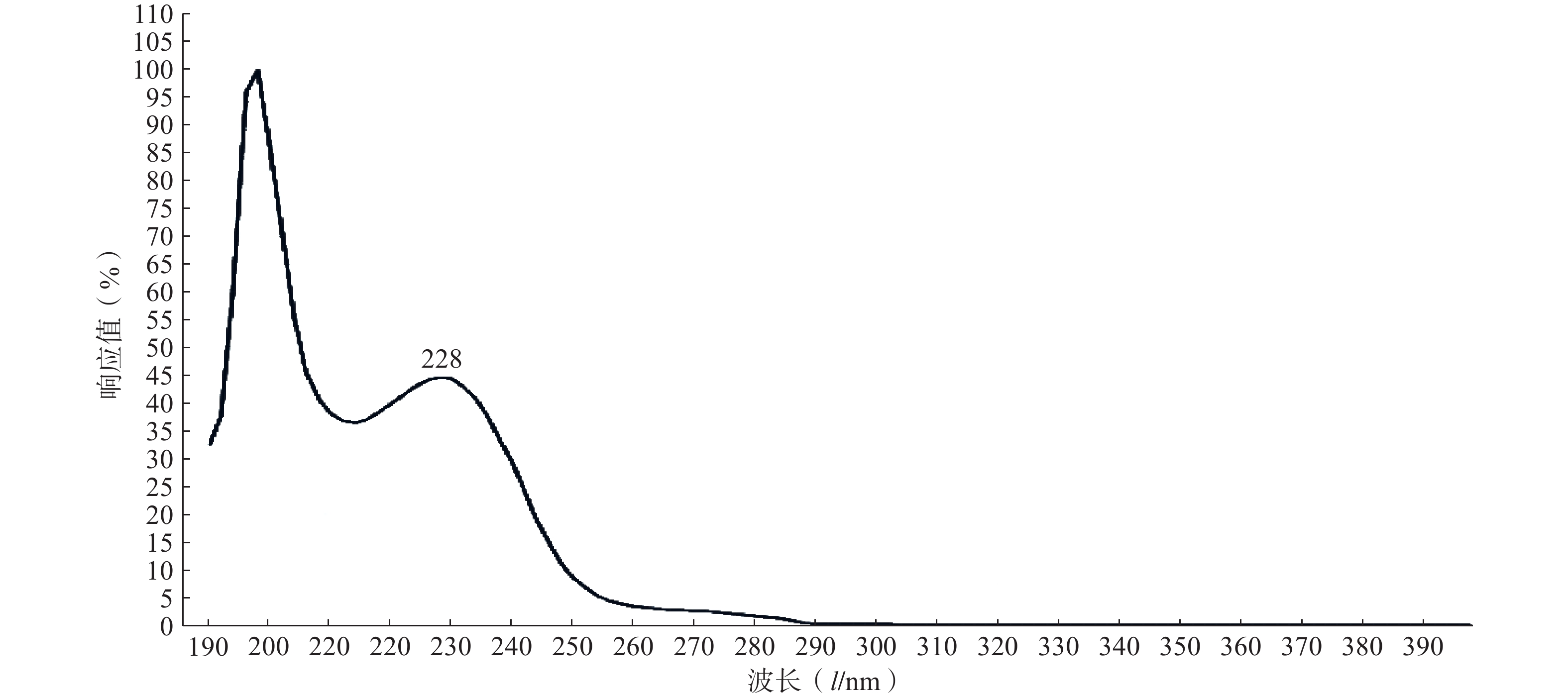
实验结果如图1所示,选用PTX-PA的最大吸收波长为228 nm为测定波长。
3.1.2 专属性考察
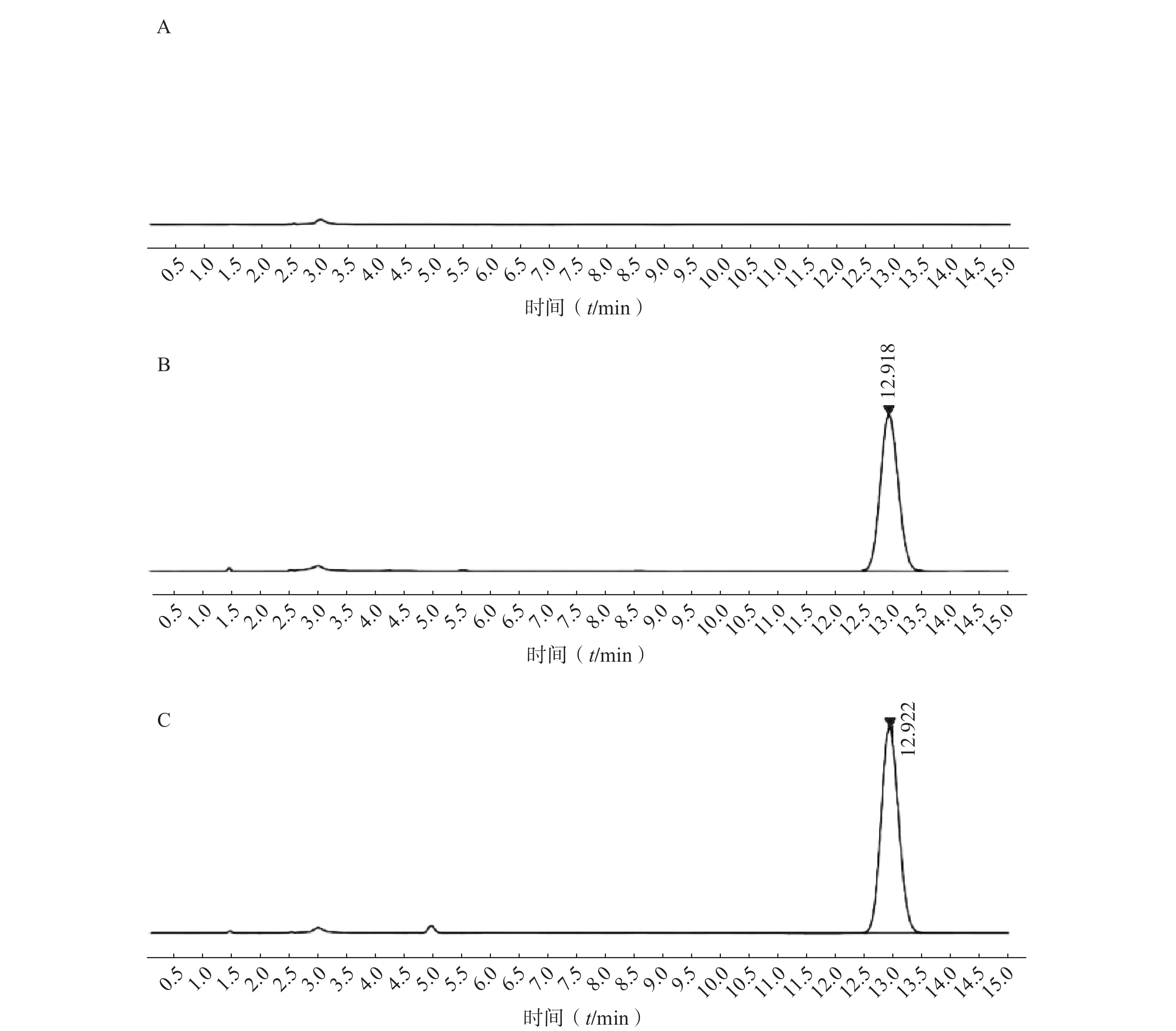
如图2所示,本章所建立的色谱条件对PTX-PA检测具有专属性,溶剂以及样品中的辅料对PTX-PA的检测不产生干扰。
3.1.3 线性关系考察
按照2.2.4方法进行回归,得PTX-PA的吸收峰面积-浓度在1~100 μg/ml的浓度范围内为线性方程:A=15.14 C+10.81(r=0.999 8)。
3.1.4 精密度考察
按照2.2.5方法进行精密度考察,结果如表1所示,日内与日间精密度各时间点峰面积的RSD均小于3%,表明仪器的日内与日间精密度符合测定要求。
表 1 PTX-PA的精密度考察结果(n=5)理论浓度(μg/ml) 实测浓度(μg/ml) RSD(%) 日内 5.00 4.98±0.13 2.65 25.00 25.30±0.55 2.18 100.00 99.66±1.11 1.11 日间 5.00 4.99±0.11 2.31 25.00 25.50±0.57 2.23 100.00 100.65±1.38 1.37 3.1.5 重复性与稳定性
按照2.2.6方法进行研究,精密吸取3种不同浓度的 PTX-PA溶液,连续进样5次并记录各吸收峰面积。各浓度峰面积RSD均<3%,表明仪器符合检测检测要求。此外,稳定性结果表明,样品溶液峰面积的RSD为0.81%,表明制备的PTX-PA溶液在24 h内稳定。
3.1.6 加样回收率
按照2.2.7方法计算加样回收率,结果如表2所示:低、中、高3个浓度的加样回收率均在95%~105%之间,且RSD分别为2.39%、1.80%、2.34%,表明本实验建立的高效液相色谱定量方法可用于PTX-PA的含量测定。
表 2 PTX-PA的加样回收率测试结果(n=5)理论浓度(μg/ml) 检测浓度(μg/ml) 回收率(%) RSD(%) 5 5.02±0.12 100.4 2.39 25 24.98±0.45 99.92 1.80 50 50.01±1.17 100.02 2.34 3.2 紫杉醇棕榈酸酯脂质体的制备及处方优化
3.2.1 PTX-PA/Lip制备方法的选择
采用不同方法制备的PTX-PA/Lip表征结果如表3所示,按照2.4方法进行统计学分析,3种制备方法的包封率无显著性差异,但采用薄膜分散法制备的PTX-PA/Lip粒径与PDI更小。因此,本研究优选薄膜分散法来构建PTX-PA/Lip。
表 3 3种常规制备方法对PTX-PA/Lip粒径、粒径分布、包封率的影响制备方法 粒径(l/nm) PDI 包封率(%) 薄膜分散法 76.76±3.39 0.104±0.02 79.38±2.00 高压均值法 125.11±5.32 0.139±0.03 78.87±2.00 挤出法 128.87±4.92 0.239±0.05 81.38±1.11 3.2.2 处方工艺筛选
(1)磷脂种类的选择
按照2.3.2(1)制备的PTX-PA/Lip表征结果如表4所示,以PC98-T为膜材制备的纳米给药系统粒径小、外观澄明、粒径分布均匀、包封率较高,因此选择PC98-T作为本研究中的磷脂。
表 4 磷脂种类对脂质体的外观形态、颗粒大小、药物包封率的影响磷脂种类 外观 粒径(l/nm) PDI 包封率(%) PC98-T 半透明 76.76±3.39 0.104±0.02 79.38±2.00 HSPC 有沉淀 177.86±5.39 0.532±0.08 59.06±1.32 EPCS 半透明 135.12±5.65 0.108±0.03 73.23±1.15 DPPC 有沉淀 158.26±4.11 0.669±0.05 53.27±2.68 (2)磷脂和药物比例的考察
按照2.3.2(2)项下确定处方中药物和磷脂的用量,其结果如表5所示,磷脂PC98-T和药物的质量比大于10时,制备的PTX-PA/Lip外观澄明度、粒子大小、粒径分散系数等参数无显著性差别。随着磷脂浓度不断增加,药物的包封率不断增加,当PC98-T和PTX-PA的质量比为20∶1时,脂质体对药物的包封率最高,后期考虑到经济成本,将PC98-T和PTX-PA的质量比定为20∶1。
表 5 磷脂和药物质量比对脂质体的外观澄明度、颗粒大小、对药物包封率的影响磷脂∶药物 外观 粒径(l/nm) PDI 包封率(%) 5∶1 略透明 134.62±2.95 0.364±0.04 58.15±1.73 10∶1 有沉淀 90.29±4.66 0.151±0.04 71.56±1.60 20∶1 半透明 84.58±1.33 0.11±0.02 83.50±0.92 30∶1 半透明 86.06±2.71 0.09±0.05 73.44±4.44 40∶1 有沉淀 88.86±1.91 0.199±0.05 68.37±11.08 (3)磷脂和胆固醇比例的考察
按照2.3.2(2)方法研究,结果如表6所示,随着胆固醇用量增多,制剂变浑浊,粒径增大,载药量显著降低。因此,胆固醇不加入本制剂的处方中。
表 6 磷脂和胆固醇质量比对脂质体外观澄明度、颗粒大小、对药物包封率的影响磷脂∶胆固醇 外观 粒径(l/nm) PDI 包封率(%) 4∶0.05 半透明 115.37±4.48 0.200±0.07 71.57±1.28 4∶0.1 半透明 160.16±3.15 0.251±0.01 61.08±3.13 4∶0.2 半透明 182.75±2.43 0.217±0.04 54.97±0.95 4∶0.3 乳白色 241.90±12.09 0.697±0.12 54.11±1.64 4∶0.4 乳白色 255.33±8.27 0.700±0.138 48.84±0.78 (4)药物和DSPE-PEG2000比例的考察
按照2.3.2(3)方法研究,其结果如表7所示,DSPE-PEG2000对包封率没有显著性影响,但当DSPE-PEG2000含量不断增加时,纳米粒子的颗粒大小先降低,当药物与DSPE-PEG2000质量比小于1∶1.5时,粒径无显著性变化,因此,药物与DSPE-PEG2000的质量比选择1∶1.5。
表 7 PTX-PA和DSPE-PEG2000的质量比对脂质体外观、粒径、药物包封率的影响药物∶DSPE-
PEG2000外观 粒径(l/nm) PDI 包封率(%) 2∶1 半透明 82.86±2.15 0.107±0.01 90.48±0.49 1∶1 半透明 78.16±2.05 0.351±0.38 90.41±0.34 1∶1.5 半透明 72.23±2.60 0.110±0.02 89.66±1.25 1∶2 半透明 74.64±1.81 0.140±0.04 90.90±2.93 1∶2.5 半透明 75.38±2.10 0.097±0.04 89.48±0.67 (5)薄膜蒸发法的温度考察
按照2.3.2(4),采用不同温度制备纳米制剂表征结果如表8所示,在筛选的5个温度中,当温度为45 ℃时,脂质体粒径最小、粒径分散性好、包封率最高,因此,本研究选用45 ℃作为薄膜蒸发温度。
表 8 温度对PTX-PA/Lip外观、粒径、包封率的影响温度(T/ ℃) 外观 粒径(l/nm) PDI 包封率(%) 35 略透明 159.42±2.42 0.545±0.08 54.94±1.85 40 半透明 105.93±6.13 0.269±0.03 73.98±1.60 45 半透明 76.97±2.50 0.105±0.049 91.13±1.45 50 半透明 91.93±2.60 0.181±0.05 80.27±2.13 55 半透明 112.23±6.37 0.233±0.06 74.15±2.12 (6)探头超声时间的考察
按照2.3.2(5)方法进行研究,结果如表9所示:处理时间较短时,纳米粒径较大,颗粒大小分布不均匀;随着超声处理的延长,粒径减小,包封率也提高;超声时间过长,脂质体结构破坏,导致药物泄露、包封率降低。因此将探头超声处理时间定为90 s。
表 9 超声处理对脂质体的外观澄明度、颗粒大小、对药物包封率的影响超声时间(t/s) 外观 粒径(l/nm) PDI 包封率(%) 30 沉淀 278.09±4.73 0.857±0.10 42.83±2.76 60 半透明 113.21±11.16 0.485±0.04 54.96±2.41 90 半透明 78.13±2.78 0.055±0.02 92.74±0.77 180 半透明 123.17±8.39 0.430±0.08 76.29±1.76 240 沉淀 261.85±4.94 0.915±0.20 50.42±2.74 3.3 紫杉醇棕榈酸酯脂质体PTX-PA/Lip的理化性质表征
综上研究,采用的最优处方和制备工艺如下:精密称取PTX-PA 20 mg、PC98-T 400 mg、DSPE-PEG2000 30 mg,加入适量二氯甲烷溶解,接着45 ℃旋蒸去除有机溶剂,再向圆底烧瓶底部薄膜中加入10 ml重蒸水(预热至同等温度),震荡、水化,得PTX-PA/Lip粗品,最后粗品探头超声(90 s)、过滤(0.22 μm),得最终样品PTX-PA/Lip纳米给药系统。
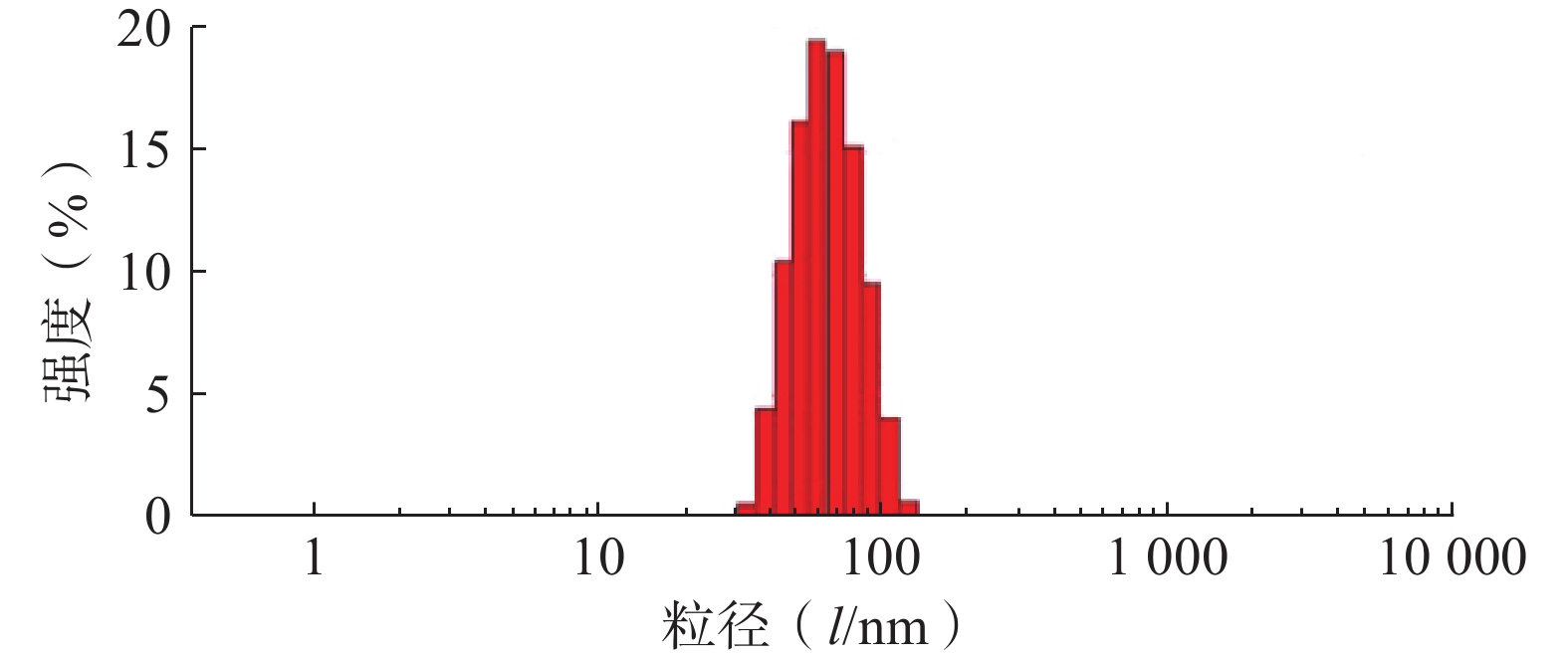
采用Zeta-sizer Nano粒度仪测定最优PTX-PA/Lip的粒径、PDI与zeta电位。结果如图3所示,制备的PTX-PA/Lip脂质体粒径大小为(62.75±1.81) nm,PDI为(0.076±0.020),Zeta电位为(−15.9±0.21) mV,表明制备的PTX-PA/Lip纳米给药系统粒径较小、分布均匀、具有良好的分散性。
4. 总结
本实验合成紫杉醇前药——紫杉醇棕榈酸酯PTX-PA,建立PTX-PA的HPLC定量测定方法,经一系列方法学验证,表明其符合PTX-PA定量分析要求,为后续试验奠定了基础。本实验采用薄膜分散法制备PTX-PA脂质体,工艺简便,技术成熟,并通过单因素筛选对PTX-PA脂质体进行处方优化。本文基于纳米技术成功制备出棕榈酸修饰的紫杉醇脂质体,增强了紫杉醇在靶细胞的递送,为PTX-PA后续的药效学研究奠定基础。
-
表 1 CuS纳米粒单因素考察结果(n=3)
因子 水平 粒子大小 (nm) 多分散指数 搅拌时间 (min) 3 18.13±5.42 0.27±0.01 5 16.12±1.29 0.27±0.01 7 15.40±1.86 0.26±0.01 水浴温度 (℃) 80 14.61±4.35 0.27±0.01 90 16.12±1.29 0.27±0.01 100 22.85±1.71 0.26±0.01 水浴时间 (min) 10 18.12±6.31 0.25±0.02 15 16.12±1.29 0.27±0.01 20 21.30±5.71 0.24±0.01 PVP浓度 (mg/ml) 80 25.56±4.40 0.27±0.01 100 16.12±1.29 0.27±0.01 120 20.72±3.38 0.25±0.02 Cu:S 3:1 15.08±2.92 0.26±0.01 2:1 20.25±1.16 0.27±0.01 1:1 16.12±1.29 0.27±0.01 1:2 11.34±3.08 0.25±0.02 1:3 11.27±2.88 0.25±0.02  下载: 导出CSV
下载: 导出CSV
表 2 星点设计因素和水平
因子 水平 −1.682 −1 0 +1 +1.682 X1 (℃) 60.00 70 85 100 110.20 X2 (mg/ml) 66.36 80 100 120 133.64 X3 0.25 0.33 1.67 3 4
下载: 导出CSV
表 3 星点设计实验安排表和结果(n=3)
序号 X1 (℃) X2 (mg/ml) X3 Y1 (nm) 1 −1 −1 −1 13.04 2 +1 −1 −1 14.23 3 −1 +1 −1 14.48 4 +1 +1 −1 13.39 5 −1 −1 +1 24.37 6 +1 −1 +1 27.30 7 −1 +1 +1 18.62 8 +1 +1 +1 20.73 9 −1.682 0 0 18.87 10 +1.682 0 0 15.62 11 0 −1.682 0 24.01 12 0 +1.682 0 17.06 13 0 0 −1.682 13.92 14 0 0 +1.682 15.11 15 0 0 0 18.81 16 0 0 0 16.84 17 0 0 0 16.14 18 0 0 0 17.89 19 0 0 0 16.20 20 0 0 0 16.30
下载: 导出CSV
-
[1] YANG J C, SHANG Y, LI Y H, et al. An “all-in-one” antitumor and anti-recurrence/metastasis nanomedicine with multi-drug co-loading and burst drug release for multi-modality therapy[J]. Chem Sci,2018,9(36):7210-7217. doi: 10.1039/C8SC02305K [2] YANG W T, GUO W S, LE W J, et al. Albumin-bioinspired Gd: CuS nanotheranostic agent for in vivo photoacoustic/magnetic resonance imaging-guided tumor-targeted photothermal therapy[J]. ACS Nano,2016,10(11):10245-10257. doi: 10.1021/acsnano.6b05760 [3] LIN X D, FANG Y, TAO Z H, et al. Tumor-microenvironment-induced all-in-one nanoplatform for multimodal imaging-guided chemical and photothermal therapy of cancer[J]. ACS Appl Mater Interfaces,2019,11(28):25043-25053. doi: 10.1021/acsami.9b07643 [4] LI W T, PENG J R, TAN L W, et al. Mild photothermal therapy/photodynamic therapy/chemotherapy of breast cancer by Lyp-1 modified Docetaxel/IR820 Co-loaded micelles[J]. Biomaterials,2016,106:119-133. doi: 10.1016/j.biomaterials.2016.08.016 [5] ZHANG Z J, WANG J, CHEN C Y. Near-infrared light-mediated nanoplatforms for cancer thermo-chemotherapy and optical imaging[J]. Adv Mater,2013,25(28):3869-3880. doi: 10.1002/adma.201301890 [6] LIAO J, LI W, PENG J, et al. Combined cancer photothermal-chemotherapy based on doxorubicin/gold nanorod-loaded polymersomes[J]. Theranostics,2015,5(4):345-56. doi: 10.7150/thno.10731 [7] WANG Y, HUANG X, TANG Y, et al. A light-induced nitric oxide controllable release nano-platform based on diketopyrrolopyrrole derivatives for pH-responsive photodynamic/photothermal synergistic cancer therapy[J]. Chemical science,2018,9(42):8103-8109. doi: 10.1039/C8SC03386B [8] LI Y, LU W, HUANG Q, et al. Copper sulfide nanoparticles for photothermal ablation of tumor cells[J]. Nanomedicine,2010,5(8):1161-71. doi: 10.2217/nnm.10.85 [9] LONGMIRE M, CHOYKE P L, KOBAYASHI H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats[J]. Nanomedicine,2008,3(5):703-717. doi: 10.2217/17435889.3.5.703 [10] PETROS R A, DESIMONE J M. Strategies in the design of nanoparticles for therapeutic applications[J]. Nat Rev Drug Discov,2010,9(8):615-627. doi: 10.1038/nrd2591 [11] ZHOU M, LI J J, LIANG S, et al. CuS nanodots with ultrahigh efficient renal clearance for positron emission tomography imaging and image-guided photothermal therapy[J]. ACS Nano,2015,9(7):7085-7096. doi: 10.1021/acsnano.5b02635 [12] PENG S W, HE Y Y, ER M, et al. Biocompatible CuS-based nanoplatforms for efficient photothermal therapy and chemotherapy in vivo[J]. Biomater Sci,2017,5(3):475-484. doi: 10.1039/C6BM00626D [13] WANG P, SHEN M Q, ZHOU H, et al. MOF-derived CuS@Cu-BTC composites as high-performance anodes for Lithium-Ion batteries[J]. Small,2019,15(47):e1903522. doi: 10.1002/smll.201903522 [14] LI X J, ZHOU J J, DONG X N, et al. In vitro and in vivo photothermal cancer therapeutic effects of gold nanorods modified with mushroom β-glucan[J]. J Agric Food Chem,2018,66(16):4091-4098. doi: 10.1021/acs.jafc.8b00292 [15] WANG R P, HE Z S, CAI P J, et al. Surface-functionalized modified copper sulfide nanoparticles enhance checkpoint blockade tumor immunotherapy by photothermal therapy and antigen capturing[J]. ACS Appl Mater Interfaces,2019,11(15):13964-13972. doi: 10.1021/acsami.9b01107 [16] GUO L R, PANDERI I, YAN D D, et al. A comparative study of hollow copper sulfide nanoparticles and hollow gold nanospheres on degradability and toxicity[J]. ACS Nano,2013,7(10):8780-8793. doi: 10.1021/nn403202w -

 点击查看大图
点击查看大图
计量
- 文章访问数: 7598
- HTML全文浏览量: 2650
- PDF下载量: 39
- 被引次数: 0
