


 下载:
下载:

 下载:
下载:

-
生物色谱法( bio-chromatography)于20世纪80年代中后期出现[1],是由生命科学与色谱分离技术交叉形成的一种极具发展潜力的新兴色谱技术。随着色谱柱制备技术和在线联用技术的深入发展,各种具有生物活性的材料,如蛋白质、细胞膜、仿生物膜、活细胞、细胞壁等纷纷被作为固定相成功投入研究。对于生物色谱技术来说,搭载生物材料的色谱固定相是技术的核心所在。对于适宜生物材料的选取、构建以及对于生物材料固定方法学的开发,使得固定相在最大程度上模拟体内的生理过程,是生物色谱技术最重要的研究方向[2-5]。
-
针对膜受体的活性分子筛选所提出的细胞膜色谱技术发展成为近年来的代表性生物色谱技术,西安交通大学贺浪冲、王嗣岑团队[6-8]开拓了该领域并进行了大量的理论和应用研究工作。该技术原理是直接提取细胞膜并固定于硅胶载体制备液相色谱固定相,通过结合受体动力学与液相色谱分析,在色谱柱内动态模拟药物在体内与受体相互作用的过程,并通过色谱参数来评价化合物的亲和活性。细胞膜色谱固定相[9-10]很好地保住了膜受体的结构和活性,且适用于与各类色谱质谱系统的联用。Ding等[11-12]在细胞膜色谱固定相方法学方面开展了大量工作,对细胞用量、破碎条件、固定相合成及填装流程进行了全面的方法学优化,对色谱柱质量标准具有较为精准的把控。此外,通过采用外源转染携带特定蛋白基因的质粒构建高表达细胞系,可实现针对特定靶受体的活性化合物筛选。但这种直接提取细胞膜并固定于载体所制备的生物色谱固定相不可避免地带来专属性和灵敏度两方面的局限性[13],主要源于细胞膜上其他膜受体的干扰和膜蛋白自身的低丰度、含量不可控等特性,不适用于靶向目标膜受体活性成分的高通量精准分析。
-
利用蛋白脂质体重构技术(proteoliposome re-constitution)[14-15]制备人工仿生膜镶嵌膜受体,包裹于硅胶基质表面,作为色谱固定相,是一种新近发展的体外模拟膜受体生物构象和微环境的新方法。其原理是将二油酰基磷脂酰胆碱、二油酰磷脂酰甘油、棕榈油酰磷脂酰胆碱、胆固醇等细胞膜含有的磷脂类成分与纯化或重组的膜受体按一定比例混合,采用不同的水化超声条件,制备成单层脂质囊泡、双层膜微胞、碟状胶束、平面磷脂膜等形式,实现膜受体-脂质体镶嵌模型重构[16-17]。膜受体脂质体是体外膜受体的理想存在形式,Mathiasen等[14]发展了两种膜受体-脂质体镶嵌模型,实现蛋白数量和融合度稳定可控,并将其用于纳米尺度的高含量分析,其研究结果发表在2014 年的《Nature Methods》上。相比较于细胞膜色谱固定相,膜受体脂质体具有蛋白种类和含量可控,磷脂和蛋白易于标记修饰等优势[18-19]。然而,此项技术仍然存在着蛋白在重构过程中发生结构改变而失活的风险。
-
分子生物色谱(MBC)是基于生物大分子特异性识别原理,属于扩展的亲和色谱[20]。广泛用于研究药物与血浆蛋白、糖蛋白、受体、DNA等大分子的相互作用关系,揭示了药物的血浆结合率等重要药理特性,而受体生物色谱[21-22]不仅能判定药物在体内的作用靶点,还可以研究药物与受体的作用强度以及与其他药物的竞争作用。对于生物色谱固定相,无论是应用于药物筛选,还是对蛋白-蛋白或蛋白-药物相互作用的研究,如何能够将大量功能完整的蛋白固定到载体表面都是技术关键。目前,色谱固定相上的蛋白固定化策略主要可分为物理吸附、化学键合、亲和标签偶联[23]。
-
物理吸附即通过蛋白质与表面之间的弱相互作用(即氢键、静电相互作用、疏水相互作用和范德华力)来实现蛋白质固定化。利用吸附原理进行偶联主要有两点好处,一是不需要对蛋白进行修饰,二是不需要额外的偶联剂[23-24]。在细胞膜色谱固定相及仿生膜生物色谱固定相发展初期,生物材料与硅胶基质的融合依赖于磷脂膜上氨基与硅胶表面硅羟基的氢键和静电吸附作用力。这种固定方式在实际应用中存在着很明显的缺陷[25],主要在于吸附作用力较弱,吸附过程可逆,且取向随机。随着时间推移,蛋白易从硅胶上脱落失活,导致色谱柱寿命短,稳定性不够[26]。
-
鉴于物理吸附的不足,化学修饰策略成为探索热潮。化学修饰策略主要可分为两种途径:①对生物分子进行化学修饰;②对固定相载体进行化学修饰。根据蛋白结构特征,可以对其天然存在的官能团进行共价修饰从而与固定相形成共价结合,以获得更加稳定的附着[24-25]。考虑到蛋白活性位点和固定相的空间相互作用,主要对蛋白质暴露的氨基酸的N端和C端进行修饰,用于修饰的基团包括氨基、羧基以及巯基等[24,27]。蛋白质与固定相载体的共价键结合又可分为非特异性结合与定点结合。绝大部分使用传统非特异性结合方法的生物大分子都存在结合取向随机的问题[28],此外,当随机固定化蛋白与表面之间的相互作用太强时,也存在变性的可能性。为了使蛋白质能够在固定相表现均匀的定向排列,开发定点结合的方法显得具有重要意义。
对固定相载体进行化学修饰,很好地避免因蛋白修饰可能影响蛋白生物活性的问题。本课题组利用固定相载体化学修饰技术,先后发展了3-氨基丙基三乙氧基硅烷(APTES)、 3-巯丙基三甲氧基硅烷(MPTS)修饰的硅胶(图1),使硅胶表面游离醛基、酯基等活性基团,与磷脂膜或蛋白上的氨基共价键合[12]。通过考察发现这种修饰方式有效提升了生物色谱柱的使用寿命和稳定性,并由此使得生物色谱在原代细胞、干细胞、纯蛋白等生物材料上的适用性更强。在固定相共价修饰的基础上,纯蛋白色谱固定相也易实现,此技术可广泛应用于各类胞膜及胞质蛋白的亲和活性成分的筛选与相互作用分析。
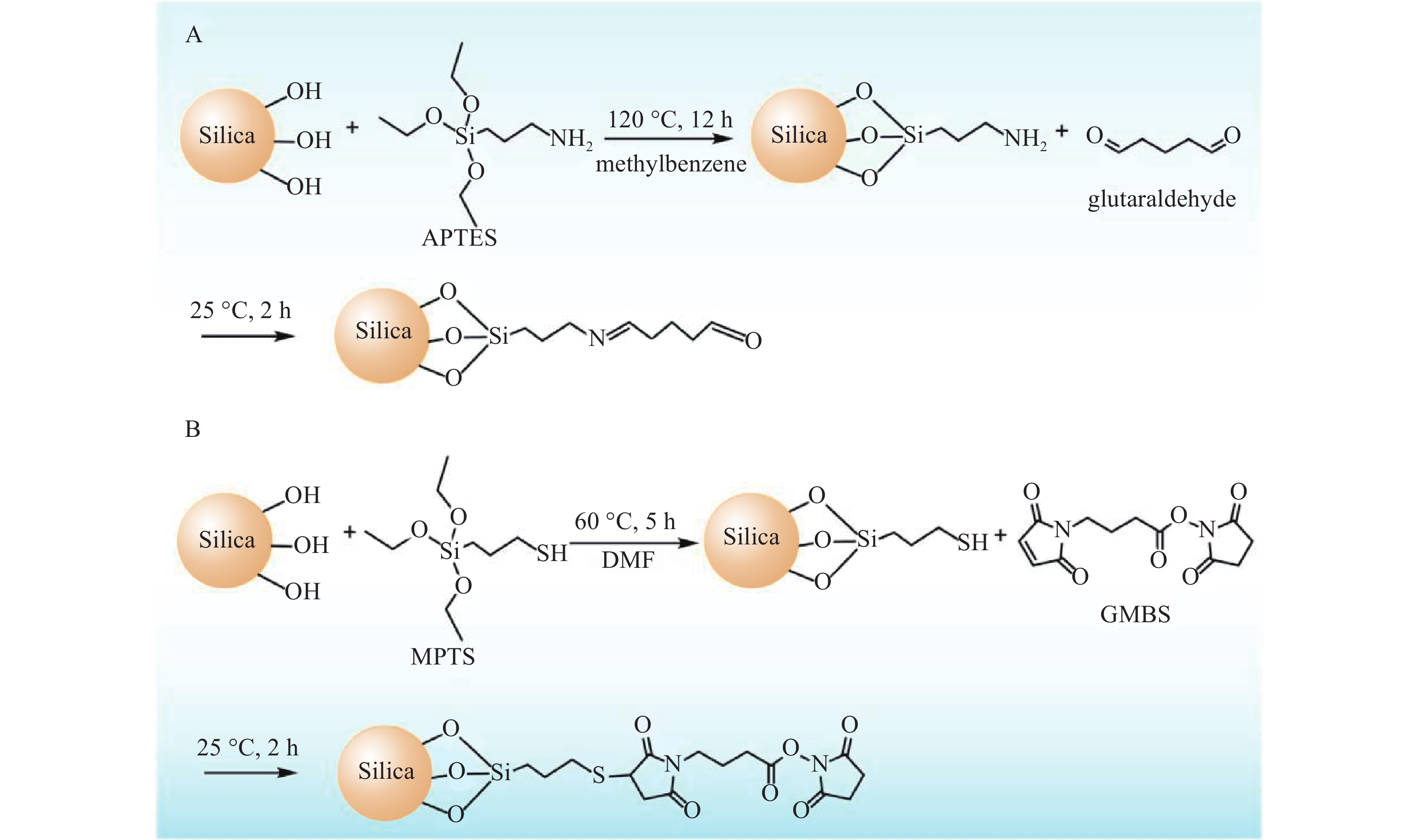
图 1 硅胶固定相的APTES(A)和MPTS(B)共价活性基团修饰合成路线
-
亲和标签偶联法则可以在更温和的条件下,使蛋白质定向均匀地连接到固定相上。不仅降低了蛋白质降解的风险,而且可以解离蛋白质,重复使用固定相[24,27]。生物素-亲和素系统稳定性很高,两者结合亲和常数可为抗原-抗体反应的百万倍,形成复合物的解离常数很小,呈不可逆反应性;而且酸、碱、变性剂等其他苛刻条件均不影响其结合[24,29-30]。Kim等[31]构建了一种基于氢醌笼状生物素表面的蛋白质模式形成方法,此方法还允许通过使用预先定型的电极阵列来选择性地产生生物素以固定目标蛋白(图2)。His是最受欢迎的标签,它是6个组氨酸残基组成的融合标签,可插入在目的蛋白的C末端或N末端,其主要优势在于分子量小,一般不影响蛋白的功能;免疫原性相对较低;纯化条件温和等[32]。
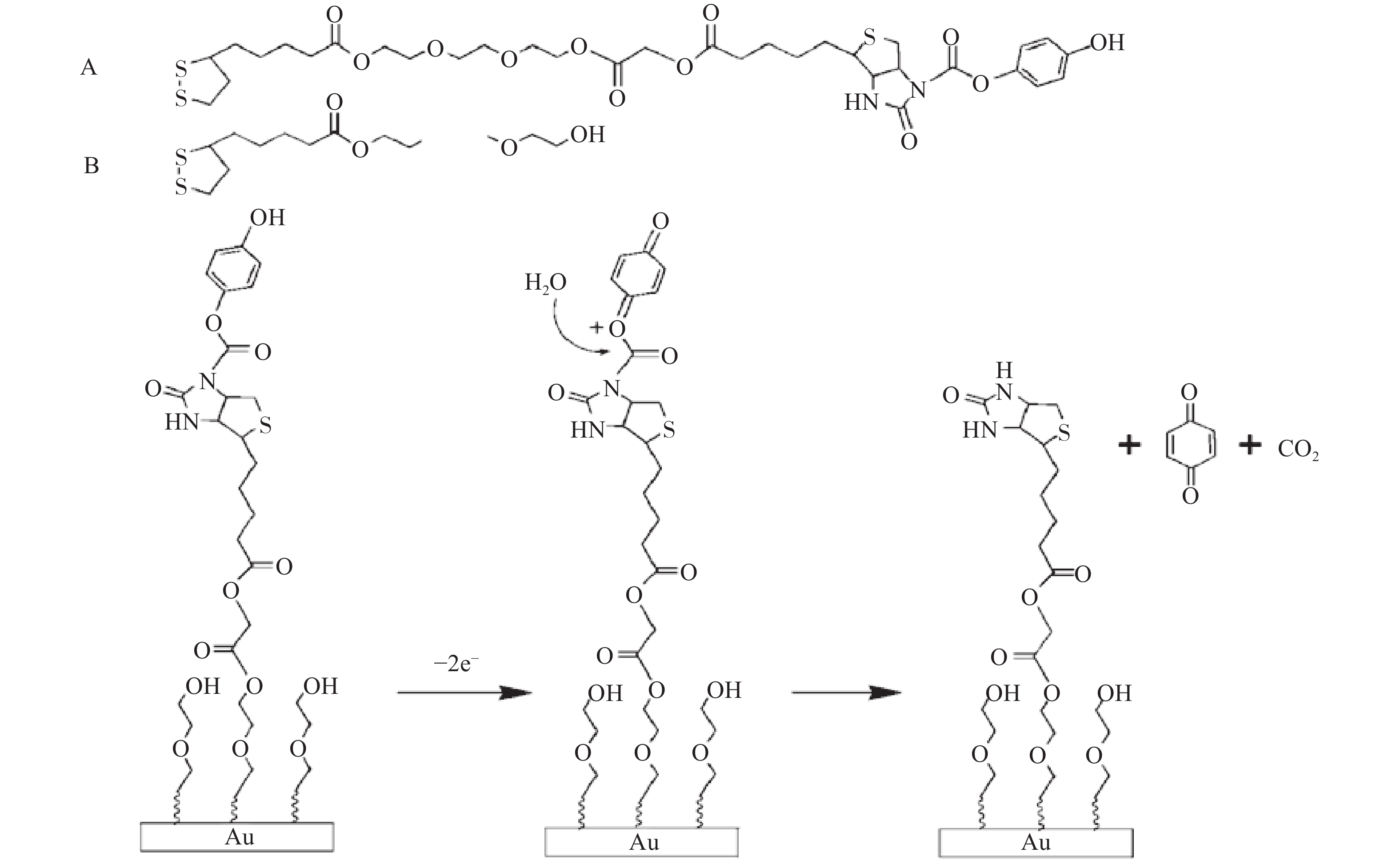
图 2 在氢醌笼状生物素修饰金表面生成生物素的电化学氧化反应原理图
-
当药物随流动相流经生物色谱,由于不同成分与固定相上的生物活性物质的作用方式及作用程度的差别而在色谱柱上表现出不同的保留特性, 以此为基础,生物色谱已广泛应用于中药等复杂体系的活性化合物筛选分析[33]。细胞膜色谱技术自提出以来,已经用于160多种植物或中药的活性成分筛选,极大地推动了中药药效物质基础的发现[34-35]。在细胞膜色谱技术的基础上,一种全二维-CMC-液相色谱-高分辨质谱联用分析系统发展起来,通过对固定相不断进行优化,结合特定膜受体的高表达细胞构建技术,固定相共价修饰技术,使得细胞膜色谱技术在自动化、高通量、稳定性与专属性各方面性能提升的同时,也拓展应用于更多珍稀生物材料的筛选分析。Chen等利用双通道全二维HepG2/CMC/TOFMS分析系统(图3),成功筛选出黄柏和苦参中的潜在活性成分[36]。Ding等采用APTES修饰硅胶策略将HepG2干细胞膜键合于固定相上,从中药丹参中筛选出了作用于肝癌干细胞膜的活性组分[12]。除此之外,固定相共价修饰技术推进了纯蛋白生物色谱分析系统的发展,使筛选系统实现针对特定受体的高特异性筛选。Gu等采用MPTS修饰硅胶固定相固定ACE2受体,从连花清瘟胶囊给药的健康人血浆中筛选出8种靶向ACE2的活性化合物,并证实其中5种化合物具有对ACE2酶活性具有较高的抑制作用,为连花清瘟胶囊临床上防治新冠肺炎提供了直接的体内药效物质基础和潜在分子机制依据[37]。

图 3 全二维细胞膜色谱分析系统示意图
-
色谱过程的本质是基于溶质、固定相与流动相之间的相互作用。当生物色谱兴起后,很快被用于生物分子之间的相互作用分析,应用范围涉及药物分子构效关系、靶点结合验证、结合解离动力学分析、蛋白关键作用位点研究等。James和Philips提出的前沿色谱法( frontal affinity chromatography)被广泛用于研究蛋白-配体间的相互作用,可同时了解药物蛋白结合情况和测定药物-蛋白平衡解离常数和生物利用度等药动学参数[38]。当色谱走向微型化后,相互作用分析从小分子药物-大分子拓展至大分子之间的作用分析,联合蛋白质组学技术,对蛋白关键作用位点的探测和构效关系研究,通过相互作用能力分析进行多肽及抗体类药物开发等前沿应用正处于快速发展阶段[39-40]。
-
生物色谱作为一种体外分析手段,在发展进程中将始终围绕着两个核心:①开发更好地模拟生物环境、适用性更佳、特异性更高的新型生物色谱固定相;②建立低生物用量、高灵敏度、功能化更全面的生物色谱分析系统。两者相互推进发展,最终建立起令人满意的分析策略。以下列举两点生物色谱分析方法学的新思路:
-
填充生物色谱柱的最大困难依然在于固定相制备困难,色谱柱性能不够稳定[41]。整体柱是一种利用有机或无机聚合方法在色谱柱内进行原位合成,形成连续床固定相的色谱柱。与填充柱相比,其优越的多孔性能、良好的重现性和机械强度使其具有更稳定的色谱行为和更高效的分离性能,因有望克服传统色谱分离介质的局限性而备受关注[42-44]。Svec教授认为[45],新型功能化整体柱的开发是当下整体柱发展的主流。随着多肽、蛋白质、DNA等各种生物大分子成功发展成为整体柱固定相材料,基于整体柱的生物色谱系统已广泛应用于复杂样品的分离纯化、小分子及抗体药物的筛选、蛋白与配体间的相互作用以及糖蛋白/磷酸化蛋白组学分析等多个生物领域[46]。近年来,暨南大学江正瑾课题组发展了仿生磷脂膜整体柱以及类磷脂膜整体柱[47],即通过在柱内键合脂质膜或柱后衍生化使得固定相表面暴露磷脂链来模拟细胞膜微环境,用于药物研发以及药物与细胞膜之间的相互作用研究[47]。总之,整体柱固定相制备过程简单,且因其优越的固定相性能,整体柱的通量与分析速度相比传统填充色谱有了很大提高,有望克服传统生物色谱固定相制备和应用中的不足,成为近年来新药研发和色谱研究领域的热点之一[43,48]。
-
新药研发、蛋白质组学分析、中药复杂成分分析等前沿领域的快速发展,对分析检测方法的灵敏度、样品通量等都提出了更高的要求。而且,对于生物色谱而言,生物材料往往获取不易,十分珍贵,常规色谱系统在生物领域的适用性上面临着很大挑战。Nano液相色谱分析系统是现阶段实现低生物用量、高灵敏度分析的重要手段[49-50],为新型生物色谱固定相的开发提供了有利条件。本课题组曾在nano色谱柱中装填细胞膜及仿生膜色谱固定相,实现了nano液相色谱串联三重飞行时间质谱用于中药活性成分的筛选分析。此外,nano液相能够更方便地与各种质谱联用,降低了对样品量的限制,将为多肽药物的筛选、生物大分子之间相互作用分析、蛋白组学分析等提供广阔的应用前景。
Advances in methodologies for preparation and analysis of new biochromatic stationary phase
-
摘要: 生物色谱法是一种极具发展潜力的新兴色谱技术,已广泛用于药物筛选及生物分子间相互作用分析。其技术核心是生物分子的色谱固定相,现今主要发展了细胞膜色谱,人工仿生膜色谱以及通过开发多种固定化策略将蛋白等直接固定于固定相载体。本文对新型生物色谱固定相方法学研究进展及基于新型固定相的生物色谱分析应用研究现状进行综述,并展望了基于整体柱的生物色谱固定相及微型生物色谱分析系统的应用前景。Abstract: Biochromatography is a new chromatographic technology with great development potential. It has been widely used in drug screening and biomolecular interaction analysis. The core of this technology is the chromatographic stationary phase of biomolecules. Nowadays, it mainly develops cell membrane chromatography, artificial biomimetic membrane chromatography and the various immobilization strategies to directly immobilizes proteins on the stationary phase carrier. This paper reviews the research progress of new biochromatographic stationary phase and the application of biochromatographic analysis based on new stationary phase. And, the applications of biochromatographic stationary phase and micro biochromatographic analysis system based on monolithic column are prospected.
-
Key words:
- biochromatography /
- immobilization method /
- drug screening /
- monolithic column
-
紫苏为重要的药食两用中药,来源于唇形科植物紫苏Perilla frutescens(L.)Britt,干燥叶与成熟果实分别作为紫苏叶与紫苏子入药,历版《中国药典》均有收载,紫苏子能降气化痰,止咳平喘,润肠通便,用于痰壅气逆,咳嗽气喘,肠燥便秘[1]。紫苏叶有解表散寒,行气和胃的功效,用于风寒感冒,咳嗽呕恶,妊娠呕吐,鱼蟹中毒。
紫苏在我国栽培极广,紫苏叶除了供药用外,还可作蔬菜、香料以及代茶。紫苏叶和肉类煮熟可增加后者的香味。日本人多用于料理,作为生鱼片佐料。紫苏子榨出的油(苏子油)也可供食用。由于紫苏应用历史悠久,形态变异大,栽培品种多,通常将叶背腹面均绿色者称为白苏,叶背为紫色者称为紫苏。紫苏与白苏的分类处理至今仍存在较大争议。如《中国植物志》[2]与《中国药典》[1]均将紫苏与白苏合为一种。而《中药志》[3]《中药辞海》[4]《新编中药志》[5]以及《中药品种理论与应用》[6]等均认为紫苏为白苏的变种。
研究表明,紫苏与白苏的形态差别并非由于栽培条件差异导致,其化学成分方面也存在明显区别,紫苏的挥发油类成分主为紫苏醛型,而白苏为紫苏酮型[7],本文对药食两用中药紫苏的名称、形态和功效进行本草考证,以正本清源,为临床安全合理应用提供科学依据。
1. 紫苏与白苏的名称考证
紫苏原名苏,始载于《名医别录》,列为中品。李时珍在《本草纲目》中也记载:“苏性舒畅,行气和血,故谓之苏”[8]。陶弘景所著的《本草集经注》记载,“苏,味辛,温。主下气,除寒中,子尤良。叶下紫色而气甚香,其无紫色不香似荏者,名野苏,不堪用”。
宋代苏颂所著的《图经本草》记载:“苏,紫苏也。谨按《尔雅》谓苏为桂荏。盖以其味辛,而形类荏,乃名之。然而苏有数种,有水苏、白苏、鱼苏、山鱼苏,皆是荏类”[9]。根据其形态描述及附图(图1),并为后来的本草文献支持,苏为现代唇形科植物紫苏Perilla frutescens (Linn.) Britt.。《救荒本草》记载:紫苏一名桂荏,又有数种,有勺苏、鱼苏、山苏,出简州及无为军,今处处有之[10]。这里提到的桂荏即紫苏,李时珍在《本草纲目》 解释到:“苏乃荏类,而味更辛如桂,故《尔雅 》谓之桂荏”。产地简州及无为军,《宋史》载:“无为军,同下州。太平兴国三年,以庐州巢县无为镇建为军,以巢、庐江二县来属。”《图经本草》有附图简州苏、无为军苏,与此也相符。
苏(舒)的名称来源于其功效。《尔雅义疏》也云:“苏之为言舒也”。紫苏则是指苏之茎叶之色为紫色。《本草求真》记载,紫苏专入肺,兼入心 、脾。背面俱紫,辛温香窜,凡风寒偶伤,气闭不利,心膨气胀,并暑湿泄泻,热闭血衄、崩淋,喉腥口臭,俱可用此调治。取其辛能入气,紫能入血,香能透外,温可暖中,使其一身舒畅,故命其名曰苏[11]。紫苏又称紫舒,相传还与东汉名医华佗有关。一次华佗在某地水边采药,无意中发现一只水獭因吃了螃蟹,难受在地上打滚,找到一种紫色的草吃了后竟安然无事。后来华佗用这种紫色的草熬汤救治了因多食螃蟹腹痛的患者。因该草紫色,患者喝后病痛解除,顿觉舒服,故称之为“紫舒”[12]。
古代本草中提到的“荏”即“白苏”,桂荏则指紫苏。李时珍《本草纲目》载:“曰紫苏者,以别白苏也。其面背皆白者即白苏,乃荏也”。《植物名实图考》中对荏也有解释,“荏,别录中品,白苏也”[13]。至于现代紫苏名称较多,紫苏又称野苏、红苏、香苏,青苏等名字。白苏又称白紫苏、青苏、臭苏等。
2. 紫苏与白苏的形态考证
紫苏与白苏形态方面的差异目前争议颇多,分类上多被作为同一种处理,认为其形态的差异是由栽培条件不同导致。然而古代本草记载以及现代研究均表明紫苏、白苏在形态与成分方面存在明显差异。《图经本草》记载:“紫苏,叶下紫色,而气甚香,夏采茎叶,秋采实。《本草纲目》曰:“紫苏,其茎方,其叶圆而有尖,四围有锯齿;肥地者面背皆紫,瘠地者面青背紫”,并有附图(图2)。据此有人认为紫苏与白苏的形态差异由于生长环境导致。《本草崇原》也记载:紫苏,其叶面青背紫,气甚辛香,开花成穗,红紫色,穗中有细子,其色黄赤,入土易生,后人于壤土莳植,面背皆紫者名家紫苏。野生瘠土者,背紫面青。《别录》首次提到野生与栽培紫苏的区别。此外,还提到一种面背皆青,气辛臭香者,为荠。一种面背皆白者,名白苏,俱不堪入药。这里提到的“荠”应为现唇形科植物荠苧Mosla grosseserrata Maxim.,又称臭苏,青白苏。而《植物名实图考》记载紫苏并有附图(图3),形态特征十分清晰,与现代文献描述的紫苏形态特征相符。
白苏又称为荏,最大区别为叶不为紫色,历代本草对此均有记载,如《本草经集注》载:“荏,状如苏,高大白色,不甚香”;而《本草图经》载:“苏有数种,有水苏、白苏、鱼苏、山鱼苏,皆为荏类。白苏,方茎圆叶,不紫,亦甚香,实亦入药。事实上无论紫苏,白苏皆含挥发油,具有特殊香气,不过紫苏味更辛如桂。《本草纲目》 :“其面背皆白者即白苏,乃荏也”。《救荒本草》记载:“(荏)苗高一二尺,茎方。叶似薄荷叶,极肥大。开淡紫色花,结穗似紫苏穗,其子如黍,其枝茎对节生”(图4)。苏与荏形态上较为相似,主要为叶面颜色差异。
3. 紫苏与白苏的功效考证
紫苏叶、紫苏梗、紫苏子均可入药,紫苏叶具有解表散寒,行气和胃的功效,紫苏梗具有理气宽中,止痛,安胎的功效。紫苏子具有降气化痰,止咳平喘,润肠通便的功效。在历代本草中均有详细记载,如《别录》:“主下气,除寒中。”《食疗本草》:“除寒热,治冷气”[14] 。《日华子本草》:“补中益气。治心腹胀满,止霍乱转筋,开胃下食,并一切冷气,止脚气,通大小肠。”《履巉岩本草》:“止金疮出血,疗痔疾,煎汤洗之”。紫苏叶入药始见于南北朝时期的《雷公炮炙论》[15]。《图经本草》记载,其茎并叶,通心经,益脾胃,煮饮尤甚,与橘皮相宜,气方中多用之。 实主上气咳逆,……,若欲宣通风毒,则单用茎,去节良。
《滇南本草》:“苏叶,发汗,解伤风头疼,定吼喘,下气,宽臌,消胀、消痰。苏子,止咳嗽,降痰,定吼喘,下气,消痰涎。” 明代《本草汇言》则记载:“紫苏,散寒气,清肺气,宽中气,下结气,化痰气,乃治气之神药也”,突出强调其治气功效。明代《本草纲目》也记载:“(紫苏)行气宽中,消痰利肺,和血,温中,止痛,定喘,安胎。其味辛,入气分,其色紫,入血分。” 清代《本经逢原》则认为紫苏可“散血脉之邪。”[16]。
关于紫苏梗的功效,明代《医学入门·本草》记载:“(紫苏)治风寒湿痹,及筋骨疼痛,脚气” [17]。明代《本草通玄》认为其“能行气安胎”。清代《本草崇原》:“主宽中行气,消饮食,化痰涎。治噎膈反胃,止心腹痛”。清代《得配本草》记载:“疏肝,利肺,理气,和血,解郁,安胎” [18]。 明代《本草蒙筌》[19]:“下诸气略缓,体虚者用宜”。
清代张志聪的《侣山堂类辩》曾有详细记载[20]:“庭前植百合、紫苏各数茎,见百合花昼开夜合,紫苏叶朝挺暮垂,因悟草木之性,感天地阴阳之气而为开阖者也,……,苏色紫赤,枝茎空通,其气朝出暮入,有如经脉之气,昼行于阳,夜行于阴,是以苏叶能发表汗者,血液之汗也(白走气分,亦走血分)。枝茎能通血脉,故易思兰先生常用苏茎通十二经之关窍,治咽膈饱闷,通大小便,止下利赤白。予亦常用香苏细茎,不切断,治反胃膈食,吐血下血,多奏奇功”。详细描述了紫苏不同部位的功效,紫苏长于解表理气之功效,紫苏叶与紫苏茎功效各有不同。
紫苏与白苏均有食用记载,《证类本草》将紫苏列为菜部中品,将荏列为菜部上品[21]。紫苏多以叶食用,《救荒本草》记载,“紫苏一名桂荏,今处处有之,苗高二尺许,茎方叶似苏子叶微小,茎叶背面皆紫色而气甚……,救饥采叶炒食,煮饮亦可,子研汁煮粥食之皆好。叶可生食与鱼作羹味佳”。白苏的食用主要为种子,“子可炒食;又研杂米作粥,甚肥美。亦可笮油用”[10]。《食物本草》也记载:“(紫苏)子,研汁煮粥长食,令人肥白身香”[22]。紫苏食用沿袭至今,许多地区仍有紫苏叶生食并与生鱼片同食习惯。
紫苏子作药用,秦、汉时期《别录》就有记载:“主下气,除寒中”。南北朝《本草经集注》记载:“苏子,主下气,与橘皮相宜同疗也”。唐代《药性论》则有不同的记载,认为苏子“主上气咳逆,治冷气及腰脚中湿风结气”。五代时期《日华子本草》则认为紫苏子“主调中,益五脏,下气,止霍乱、呕吐、反胃,补虚劳,肥健人,利大小便,破癥结,消五膈,止嗽,润心肺,消痰气”。北宋时期《本草衍义》记载紫苏子“治肺气喘急”[23]。《本草纲目》认为“苏子与叶同功,发散风气宜用叶,清利上下则宜用子也”。
紫苏以种子、叶、梗入药,白苏多以种子、叶和根入药。白苏又称南苏,叶功效与紫苏相似,而白苏根又有洗疮祛风的功效。《别录》记载:“主治欬逆,下气,温中”。《日华子本草》记载:“调气,润心肺,长肌肤,益颜色,消宿食,止上气咳嗽,去狐臭,敷蛇咬”。《滇南本草》则记载:“南苏,治伤寒发热,无汗头痛,其效如神。此草治一切风寒,痰涌结而霍乱转筋,咳嗽吐痰、小儿风症,定痛止喘。梗能补中益气。根能洗疮祛风。子能开胃健脾”。
4. 紫苏的毒性与副作用
紫苏的毒副作用记载较少,多数不良反应为药物或食物的配伍导致。如《本草纲目》记载:“不可同鲤鱼食,生毒疮”。明代的《神农本草经疏》[24]:“病属阴虚,因发寒热或恶寒及头痛者,慎毋投之,以病宜敛宜补故也。火升作呕者,亦不宜服,惟可用子”。《本草通玄》则记载:“久服泄人真气”。《药性切用》提到“气虚者禁用”。《医学入门·本草》也提到“脾胃气虚常泄者禁用”。《本经逢原》记载:“性主疏泄,气虚久嗽,阴虚喘逆,脾虚便溏者皆不可用”。因紫苏性温,主下气,故阴虚、气虚及温病患者慎用。
我们对紫苏与白苏的形态与化学成分研究结果表明,白苏的挥发油类成分主要为紫苏酮型,具有一定的毒性,存在安全隐患,因此作为食用以紫苏为好,不宜使用白苏。
5. 小结
通过对紫苏名称的文献考证结果表明,历代本草所言“紫苏”“苏”“白苏”均属“荏”类,为唇形科紫苏Perilla frutescens及其变种,“紫苏”源于“苏”,为其形态与功效的延伸。李时珍区分“紫苏”与“白苏”,白苏又称为荏,紫苏又称桂荏。通常将叶背腹面均绿色者称为白苏,叶背为紫色称为紫苏。白苏不甚香,不堪入药。紫苏茎、叶、子均入药,主下气,治风寒。
根据历代本草记载,紫苏和白苏为两种不同的植物,现代分类处理尚存在争议,认为紫苏是同种植物因环境不同产生的变异。然而其形态、功效及化学成分均存在明显差别。根据历代草本的紫苏附图、形态描述,紫苏与白苏应作为不同的分类单元。
紫苏茎、叶、子均入药,功效有所不同,对于不同病症注意合理的配伍,以免产生不良反应。此外,气虚、阴虚及温病患者慎用。紫苏作为药食两用的种类,应用较为广泛,为保证临床应用的安全有效,应与白苏加以区分。
-
[1] CLONIS Y D. Affinity chromatography matures as bioinformatic and combinatorial tools develop[J]. J Chromatogr A,2006,1101(1-2):1-24. doi: 10.1016/j.chroma.2005.09.073 [2] LI Z, RODRIGUEZ E, AZARIA S, et al. Affinity monolith chromatography: a review of general principles and applications[J]. Electrophoresis,2017,38(22-23):2837-2850. doi: 10.1002/elps.201700101 [3] BATISTA-VIERA F, JANSON J C, CARLSSON J. Affinity chromatography[J]. Methods Biochem Anal,2011,54:221-258. [4] MUHAMMAD S, HAN S L, XIE X Y, et al. Overview of online two-dimensional liquid chromatography based on cell membrane chromatography for screening target components from traditional Chinese medicines[J]. J Sep Sci,2017,40(1):299-313. doi: 10.1002/jssc.201600773 [5] ZHANG H, WU Z Y, YANG Y Y, et al. Recent applications of immobilized biomaterials in herbal analysis[J]. J Chromatogr A,2019,1603:216-230. doi: 10.1016/j.chroma.2019.06.059 [6] 贺浪冲, 耿信笃. 细胞膜受体色谱法一研究药物与受体作用的新方法[J]. 生物医药色谱新进展. 1996, 3: 8-9. [7] HE L C, WANG S C, YANG G D, et al. Progress in cell membrane chromatography[J]. Drug Discov Ther,2007,1(2):104-107. [8] 贺浪冲, 杨广德, 耿信笃. 固定在硅胶表面细胞膜的酶活性及其色谱特性[J]. 科学通报, 1999, 44(6):632-637. doi: 10.3321/j.issn:0023-074X.1999.06.013 [9] HOU X F, WANG S C, ZHANG T, et al. Recent advances in cell membrane chromatography for traditional Chinese medicines analysis[J]. J Pharm Biomed Anal,2014,101:141-150. doi: 10.1016/j.jpba.2014.05.021 [10] LI M, HOU X F, ZHANG J, et al. Applications of HPLC/MS in the analysis of traditional Chinese medicines[J]. J Pharm Anal,2011,1(2):81-91. doi: 10.1016/S2095-1779(11)70015-6 [11] DING X, CHEN X F, CAO Y, et al. Quality improvements of cell membrane chromatographic column[J]. J Chromatogr A,2014,1359:330-335. doi: 10.1016/j.chroma.2014.07.071 [12] DING X, CAO Y, YUAN Y F, et al. Development of APTES-decorated HepG2 cancer stem cell membrane chromatography for screening active components from Salvia miltiorrhiza[J]. Anal Chem,2016,88(24):12081-12089. doi: 10.1021/acs.analchem.6b02709 [13] 张月华, 蒋才武. 抗肿瘤中药药效物质筛选与辨识的研究方法进展[J]. 中南药学, 2020, 18(9):1517-1522. [14] MATHIASEN S, CHRISTENSEN S M, FUNG J J, et al. Nanoscale high-content analysis using compositional heterogeneities of single proteoliposomes[J]. Nat Methods,2014,11(9):931-934. doi: 10.1038/nmeth.3062 [15] SEREBRYANY E, ZHU G A, YAN E C Y. Artificial membrane-like environments for in vitro studies of purified G-protein coupled receptors[J]. Biochim Biophys Acta,2012,1818(2):225-233. doi: 10.1016/j.bbamem.2011.07.047 [16] GARNI M, THAMBOO S, SCHOENENBERGER C A, et al. Biopores/membrane proteins in synthetic polymer membranes[J]. Biochim Biophys Acta Biomembr,2017,1859(4):619-638. doi: 10.1016/j.bbamem.2016.10.015 [17] JØRGENSEN I L, KEMMER G C, POMORSKI T G. Membrane protein reconstitution into giant unilamellar vesicles: a review on current techniques[J]. Eur Biophys J,2017,46(2):103-119. doi: 10.1007/s00249-016-1155-9 [18] LIU G Y, HOU S L, TONG P H, et al. Liposomes: preparation, characteristics, and application strategies in analytical chemistry[J]. Crit Rev Anal Chem,2020:1-21. [19] SFORZI J, PALAGI L, AIME S. Liposome-based bioassays[J]. Biology (Basel),2020,9(8):E202. [20] 郭明, 由业诚, 孔亮, 等. 分子生物色谱及其在药物筛选中的应用[J]. 大连大学学报, 2003, 24(2):38-41. doi: 10.3969/j.issn.1008-2395.2003.02.012 [21] CHEN X F, WU Y L, CHEN C, et al. Identifying potential anti-COVID-19 pharmacological components of traditional Chinese medicine Lianhuaqingwen capsule based on human exposure and ACE2 biochromatography screening[J]. Acta Pharm Sin B,2021,11(1):222-236. doi: 10.1016/j.apsb.2020.10.002 [22] QV X Y, JIANG J G, PIAO J H. Pharmacodynamic studies of Chinese medicine at levels of whole animal, cell and molecular models[J]. Curr Med Chem,2010,17(36):4521-4537. doi: 10.2174/092986710794182926 [23] STEEN REDEKER E, TA D T, CORTENS D, et al. Protein engineering for directed immobilization[J]. Bioconjug Chem,2013,24(11):1761-1777. doi: 10.1021/bc4002823 [24] RUSMINI F, ZHONG Z Y, FEIJEN J. Protein immobilization strategies for protein biochips[J]. Biomacromolecules,2007,8(6):1775-1789. doi: 10.1021/bm061197b [25] WONG L S, KHAN F, MICKLEFIELD J. Selective covalent protein immobilization: strategies and applications[J]. Chem Rev,2009,109(9):4025-4053. doi: 10.1021/cr8004668 [26] 王晓宇, 陈啸飞, 顾妍秋, 等. 细胞膜色谱研究进展及其在中药活性成分筛选中的应用[J]. 分析化学, 2018, 46(11):1695-1702. doi: 10.11895/j.issn.0253-3820.171287 [27] SANGHVI M, MOADDEL R, WAINER I W. The development and characterization of protein-based stationary phases for studying drug-protein and protein-protein interactions[J]. J Chromatogr A,2011,1218(49):8791-8798. doi: 10.1016/j.chroma.2011.05.067 [28] CHA T, GUO A, ZHU X Y. Enzymatic activity on a chip: the critical role of protein orientation[J]. PROTEOMICS,2005,5(2):416-419. doi: 10.1002/pmic.200400948 [29] 刘石锋, 陈倩, 洪广成, 等. 生物素-亲和素系统的应用研究进展[J]. 生物技术, 2018, 28(5):503-507. [30] WILCHEK M, BAYER E A. The avidin-biotin complex in immunology[J]. Immunol Today,1984,5(2):39-43. doi: 10.1016/0167-5699(84)90027-6 [31] KIM K, YANG H, JON S, et al. Protein patterning based on electrochemical activation of bioinactive surfaces with hydroquinone-caged biotin[J]. J Am Chem Soc,2004,126(47):15368-15369. doi: 10.1021/ja0459330 [32] PINA A S, BATALHA Í L, DIAS A M G C, et al. Affinity tags in protein purification and peptide enrichment: an overview[J]. Methods Mol Biol,2021,2178:107-132. [33] 周思维, 邱瑞琪, 高彩芳, 等. 生物色谱用于中药活性成分筛选[J]. 中国医药工业杂志, 2017, 48(12):1692-1697. [34] HOU X F, ZHOU M Z, JIANG Q, et al. A vascular smooth muscle/cell membrane chromatography-offline-gas chromatography/mass spectrometry method for recognition, separation and identification of active components from traditional Chinese medicines[J]. J Chromatogr A,2009,1216(42):7081-7087. doi: 10.1016/j.chroma.2009.08.062 [35] FU J, LV Y N, JIA Q Q, et al. Dual-mixed/CMC model for screening target components from traditional Chinese medicines simultaneously acting on EGFR & FGFR4 receptors[J]. Talanta,2019,192:248-254. doi: 10.1016/j.talanta.2018.09.053 [36] CHEN X F, CAO Y, LV D Y, et al. Comprehensive two-dimensional HepG2/cell membrane chromatography/monolithic column/time-of-flight mass spectrometry system for screening anti-tumor components from herbal medicines[J]. J Chromatogr A,2012,1242:67-74. doi: 10.1016/j.chroma.2012.04.034 [37] CHEN C, YANG F Q, ZUO H L, et al. Applications of biochromatography in the screening of bioactive natural products[J]. J Chromatogr Sci,2013,51(8):780-790. doi: 10.1093/chromsci/bmt002 [38] KASAI K. Frontal affinity chromatography: an excellent method of analyzing weak biomolecular interactions based on a unique principle[J]. Biochim Biophys Acta Gen Subj,2021,1865(1):129761. doi: 10.1016/j.bbagen.2020.129761 [39] LINGG N, ÖHLKNECHT C, FISCHER A, et al. Proteomics analysis of host cell proteins after immobilized metal affinity chromatography: influence of ligand and metal ions[J]. J Chromatogr A,2020,1633:461649. doi: 10.1016/j.chroma.2020.461649 [40] ZHANG Y Y, FONSLOW B R, SHAN B, et al. Protein analysis by shotgun/bottom-up proteomics[J]. Chem Rev,2013,113(4):2343-2394. doi: 10.1021/cr3003533 [41] UNGER K K, SKUDAS R, SCHULTE M M. Particle packed columns and monolithic columns in high-performance liquid chromatography-comparison and critical appraisal[J]. J Chromatogr A,2008,1184(1-2):393-415. doi: 10.1016/j.chroma.2007.11.118 [42] BUNCH D R, WANG S H. Applications of monolithic columns in liquid chromatography-based clinical chemistry assays[J]. J Sep Sci,2011,34(16-17):2003-2012. doi: 10.1002/jssc.201100189 [43] STANIAK M, WÓJCIAK M, SOWA I, et al. Silica-based monolithic columns as a tool in HPLC-an overview of application in analysis of active compounds in biological samples[J]. Molecules,2020,25(14):E3149. doi: 10.3390/molecules25143149 [44] GUO J L, LIN H, WANG J C, et al. Recent advances in bio-affinity chromatography for screening bioactive compounds from natural products[J]. J Pharm Biomed Anal,2019,165:182-197. doi: 10.1016/j.jpba.2018.12.009 [45] SVEC F, LV Y Q. Advances and recent trends in the field of monolithic columns for chromatography[J]. Anal Chem,2015,87(1):250-273. doi: 10.1021/ac504059c [46] CHEN M L, LI L M, YUAN B F, et al. Preparation and characterization of methacrylate-based monolith for capillary hydrophilic interaction chromatography[J]. J Chromatogr A,2012,1230:54-60. doi: 10.1016/j.chroma.2012.01.065 [47] ZHAO X L, CHEN W J, ZHOU Z Y, et al. Preparation of a biomimetic polyphosphorylcholine monolithic column for immobilized artificial membrane chromatography[J]. J Chromatogr A,2015,1407:176-183. doi: 10.1016/j.chroma.2015.06.056 [48] 彭坤, 吴慧慧, 李林, 等. 功能化聚合物基质整体色谱柱的研究进展[J]. 分析测试学报, 2018, 37(10):1158-1165. doi: 10.3969/j.issn.1004-4957.2018.10.006 [49] MEJÍA-CARMONA K, MACIEL E V S, LANÇAS F M. Miniaturized liquid chromatography applied to the analysis of residues and contaminants in food: a review[J]. Electrophoresis,2020,41(20):1680-1693. doi: 10.1002/elps.202000019 [50] HARA T, IZUMI Y, HATA K, et al. Performance of small-domain monolithic silica columns in nano-liquid chromatography and comparison with commercial packed bed columns with 2 µm particles[J]. J Chromatogr A,2020,1616:460804. doi: 10.1016/j.chroma.2019.460804 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 7454
- HTML全文浏览量: 3161
- PDF下载量: 70
- 被引次数: 0
