

 下载:
下载:
 下载:
下载:

-
白念珠菌是一种条件致病真菌,在健康人体口腔或胃肠道中以无害共生菌的形式存在。在免疫功能低下的患者中,能够导致危及生命的全身性感染。近年来,随着癌症患者、器官移植接受者等免疫功能低下人群不断增加,白念珠菌感染引发的严重疾病的发病率不断攀升[1-2],死亡率粗略估计已高于40%[3]。
高适应性是白念珠菌传播和致病的关键因素之一,包括对宿主和外界环境的适应性、形态的转换以及对抗真菌药物的适应性。代谢的改变能够影响白念珠菌的药物敏感性,Ene等[4]发现培养基中碳源由葡萄糖转变为乳酸后白念珠菌对抗真菌药物(咪康唑、两性霉素B、卡泊芬净)的敏感性发生改变。线粒体作为重要的代谢细胞器, 其中的某些基因缺失或者功能缺陷,会导致白念珠菌药物敏感性的变化,Sun等[5]发现线粒体复合体I相关基因GOA1和NDH51的缺失会导致白念珠菌对唑类药物敏感性上升,Edwina等[6]的研究发现线粒体关键基因FZO1的缺失能够增强白念珠菌对唑类药物的敏感性。
本课题组前期发现SDH2基因缺失导致白念珠菌致病力显著下降,并发现白念珠菌无法利用非发酵碳源[7]。生物信息学分析显示SDH2基因编码琥珀酸脱氢酶(SDH)的铁硫亚基,琥珀酸脱氢酶在三羧酸循环和线粒体电子传递链中均发挥作用,是能量代谢中重要的一环。SDH2基因可能在白念珠菌代谢过程中发挥重要的作用,那么它是否能够通过影响代谢从而改变白念珠菌的环境适应性呢?本研究聚焦SDH2基因对白念珠菌环境适应性的影响,包括对外界压力应答和药物敏感性的影响,并探索其可能的机制。
-
挑取少量冻存于−80℃、30% 甘油中的菌株,在YPD 液体培养基中活化,30℃,200 r/min振荡培养24 h后,吸取10 μl置于新的1 ml YPD液体培养基中,继续在 30℃,200 r/min振荡条件下培养 16 h,用 SDA 固体培养基划板,30℃培养 48 h,置于 4℃ 保存备用。实验时挑取 SDA 平板上的单克隆菌落置于1 ml YPD 液体培养基中,30℃,200 r/min振荡培养 16 h 过夜,使其处于对数生长期用于后续实验。
-
白念珠菌活化16 h ,用YPD液体培养基调整菌浓度至3×106细胞/ml。制备每种菌株的系列稀释液,以10倍倍比稀释成5个浓度梯度,每种白念珠菌的系列浓度稀释物各取5 μl,点在事先加入了待考察的不同浓度的化学试剂和药物的YPD琼脂平板上,倒置于孵箱内,25、30、37 ℃培养24~48 h,期间观察各菌株之间生长的差异并拍照记录。
-
收集处于对数生长期的白念珠菌细胞悬浮液,用无菌PBS洗涤3次。随后将细胞重悬于无菌PBS中调整其浓度至5×107细胞/ml,孵育2 h以耗尽能量,并加入终浓度为10 μmol/L的罗丹明6G。30℃、200 r/min振荡培养细胞悬浮液60 min,使罗丹明6G积累。洗涤白念珠菌细胞悬浮液3次,注意要保证白念珠菌细胞最终浓度为5×107细胞/ml。然后加入终浓度为20 mmol/L的葡萄糖。分别在25 、30、37℃ 条件下,200 r/min振荡培养白念珠菌细胞悬浮液2 h,取白念珠菌细胞样品(约1 ml)离心,收集上清液,在527 nm处测量吸光度。
-
使用GraphPad Prism软件对全部数据进行分析,时序检验 (Mantel Cox) 方法比较各组差异 ,当P<0.05时,表示差异具有统计学意义。
-
我们利用多种刺激剂考察了SDH2在白念珠菌应激反应中的作用。实验应用了sdh2Δ/Δ敲除菌、野生型菌和回复菌,考察在存在应激刺激剂的培养基中sdh2Δ/Δ敲除菌的生长情况与野生型菌和回复菌的区别,从而判断SDH2在白念珠菌应激反应中的作用。应激刺激剂包括细胞壁应激刺激剂(十二烷基硫酸钠和咖啡因)、渗透压应激刺激剂(氯化钠)、氧化应激刺激剂(过氧化氢、二酰胺和甲萘醌)。敏感性实验在25、30、37 ℃三种不同的温度下进行,将不同浓度相同体积的白念珠菌接种于琼脂培养板上,同样的培养板制备3份,白念珠菌生长形成明显的菌落后拍照。结果显示,与野生型菌或回复菌相比,sdh2Δ/Δ敲除菌于25 ℃在0.03%十二烷基硫酸钠培养板上生长得多(图1A)。此外,在所有培养温度下,sdh2Δ/Δ敲除菌在含咖啡因、二酰胺或甲萘醌的培养板上也较野生型菌/回复菌生长得多 (图1A、1B、1C)。

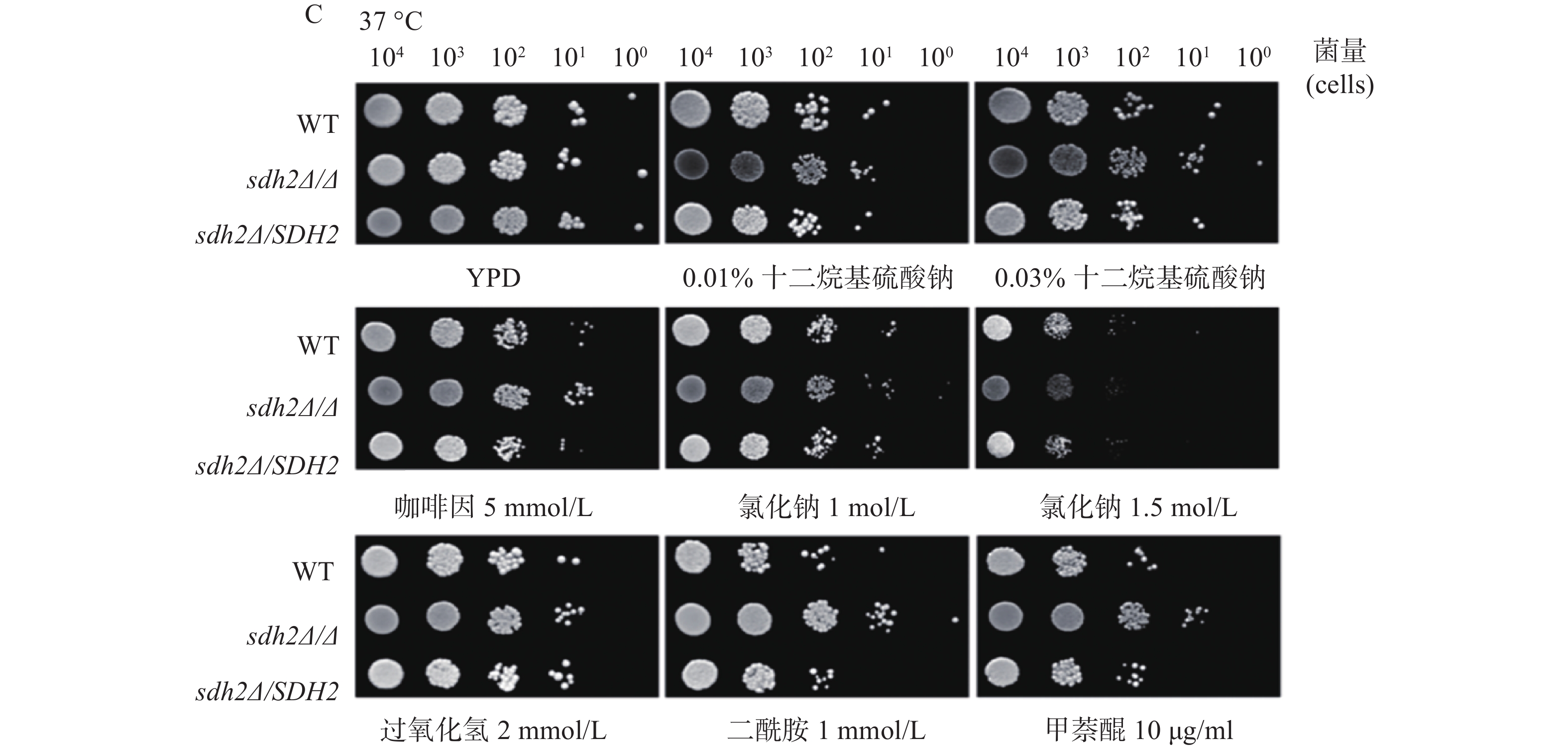
图 1 SDH2基因缺失菌与野生型菌株对各种应激刺激剂的敏感性
-
我们考察了sdh2Δ/Δ敲除菌、野生型菌和回复菌对抗真菌药物的敏感性,考察的药物包括唑类抗真菌药物特比萘芬、两性霉素B、氟胞嘧啶和阿尼芬净。将不同浓度相同体积的白念珠菌接种于琼脂培养板上,同样的培养板制备3份,白念珠菌生长形成明显的菌落后拍照。药物敏感性实验结果显示,与野生型菌/回复菌相比,sdh2Δ/Δ敲除菌在含有氟康唑、酮康唑和伊曲康唑的培养板上生长得明显减少(图2A、2B、2C),而在含有其他抗真菌药物,如特比萘芬、两性霉素B、氟胞嘧啶和阿尼芬净的培养板上没有明显差别。

图 2 SDH2基因缺失菌与野生型菌株对抗真菌药物的敏感性
-
罗丹明6G是ABC(ATP-binding cassette)转运蛋白的特异性底物,在能量不足时以被动扩散的方式进入白念珠菌细胞,加入葡萄糖供能后,可通过药物外排泵以主动转运的方式泵出细胞外。我们通过罗丹明6G的外排实验考察了SDH2敲除对白念珠菌药物外排能力的影响。结果显示在30 ℃和37 ℃ 培养温度下,通过添加葡萄糖以启动能量代谢2 h后,与野生型菌相比,从sdh2Δ/Δ敲除菌的细胞中泵出的罗丹明6G显著减少(P<0.01,见图3)。有趣的是,在25 ℃ 的培养温度下,3种菌株泵出的罗丹明6G都较30 ℃和37 ℃条件下有所减少,但各菌株之间没有显著差异(图3)。
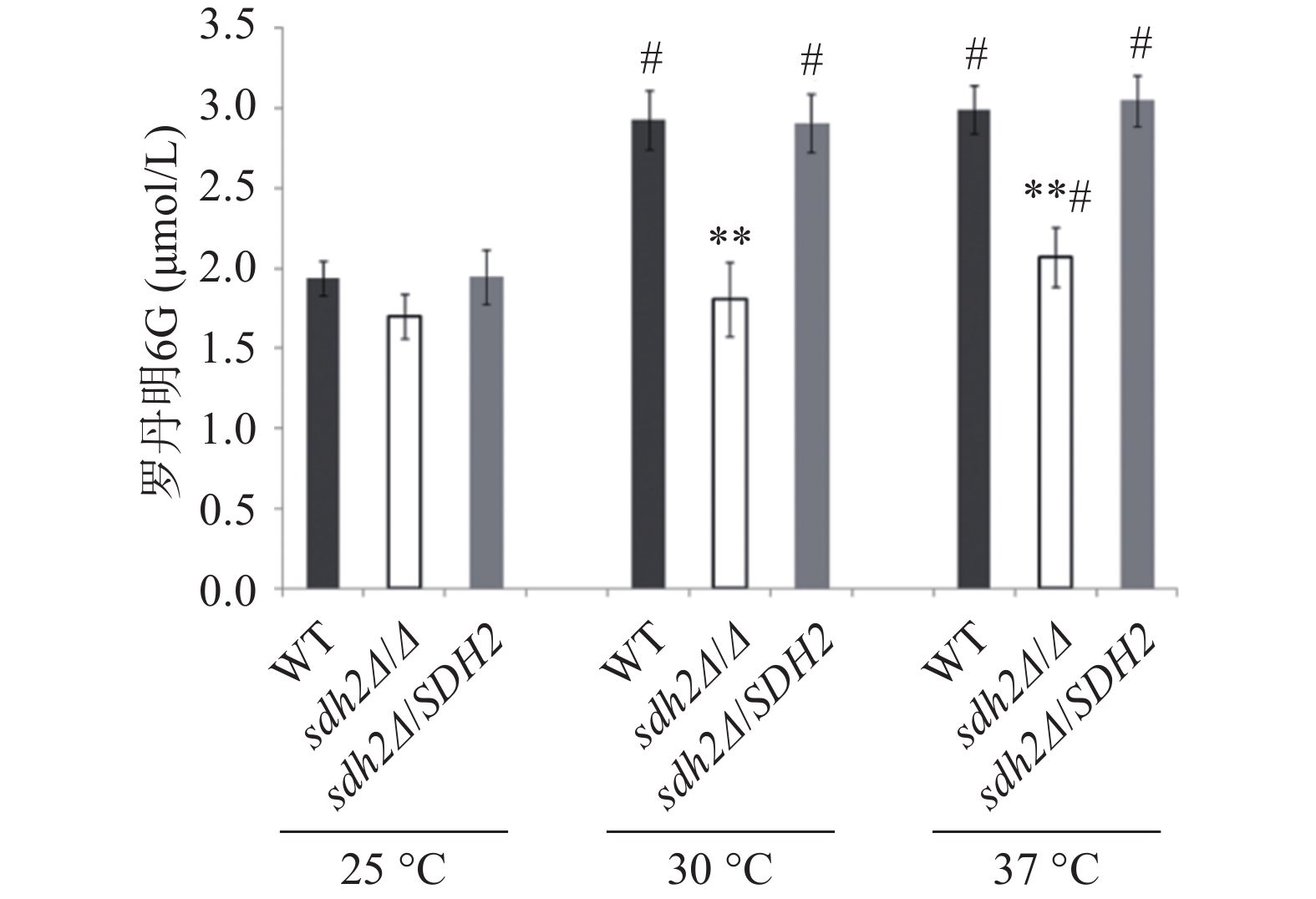
图 3 SDH2基因缺失菌与野生型菌株对罗丹明6G的外排能力
-
SDH2编码琥珀酸脱氢酶中的一个亚基,琥珀酸脱氢酶参与三羧酸循环,并且它位于线粒体内膜上,发挥电子传递的作用,对于能量代谢十分重要[8]。我们对SDH2基因在白念珠菌环境适应性中的作用进行了初步探索,实验选择在30 ℃(白念珠菌最适宜生长温度)、37 ℃(宿主温度)和25 ℃(室温)三种温度下进行。实验之所以选用三种温度,一方面是由于在不同环境温度下白念珠菌的代谢可能不同,选用不同的温度可以考察温度造成的差别;另一方面在三种温度下考察SDH2基因的功能,结果相互之间可以比较或者印证,增加信息量,有利于分析SDH2基因的功能。有意思的是SDH2基因缺失后菌株对唑类抗真菌药物(氟康唑、酮康唑、伊曲康唑)的敏感性显著上升,而对于其他类型的抗真菌药物包括丙烯胺类特比萘芬、多烯类两性霉素B、核苷类氟胞嘧啶和棘白菌素类阿尼芬净,其敏感性和野生型菌没有差异。唑类药物的敏感性与药物外排泵的功能密切相关,药物外排功能障碍可导致对唑类抗真菌药物敏感性增加[9]。药物外排泵包括ABC转运蛋白超家族和主要易化因子超家族,其中ABC转运蛋白中的Cdr1p、Cdr2p发挥作用都需要消耗能量。罗丹明6G是能量依赖型ABC转运蛋白的特异性底物,通过外排泵以能量依赖的主动转运方式排出细胞外。罗丹明6G外排实验的结果能显示能量依赖型ABC转运蛋白的功能。研究发现SDH2缺失后白念珠菌在30 ℃和37 ℃时外排能力都显著降低。SDH2对能量代谢非常重要,SDH2基因缺失后,菌株能量代谢异常,能量依赖型ABC转运蛋白的功能下降,缺失菌对唑类抗真菌药物的敏感性增加。
SDH2基因缺失后白念珠菌对咖啡因、二酰胺和甲萘醌都表现出轻微的耐受,这提示SDH2基因在白念珠菌环境应激中有一定的作用,这些环境因素包括细胞壁压力的刺激以及氧化刺激,具体的作用机制还有待进一步探讨。
综上所述,本研究发现SDH2基因缺失会导致白念珠菌对环境压力应激反应的改变;此外,SDH2基因缺失后菌株的药物外排能力降低,菌株对唑类抗真菌药物的敏感性增加。代谢对白念珠菌的环境适应能力和致病力十分重要,SDH2基因缺失导致白念珠菌对唑类抗真菌药物专特性地敏感,以SDH2为靶基因,开发真菌特异性SDH2抑制剂,有望发现与唑类药物协同的新型抗真菌药物。
The role of SDH2 gene in the environmental adaptability of Candida albicans
-
摘要:
目的 探究SDH2基因在白念珠菌环境适应性中的作用。 方法 以野生型白念珠菌SC5314、SDH2基因敲除菌sdh2 Δ/Δ 、基因回复菌sdh2 Δ /SDH2为实验对象;应用点板实验考察野生型菌、SDH2缺失菌和回复菌对外界压力刺激剂和抗真菌药物的敏感性;采用罗丹明6G外排实验考察SDH2基因缺失对白念珠菌药物外排能力的影响。 结果 SDH2基因缺失后白念珠菌对细胞壁应激刺激剂咖啡因、氧化应激刺激剂二酰胺和甲萘醌表现出轻微耐受,值得注意的是SDH2基因敲除菌sdh2 Δ/Δ 对唑类抗真菌药物的敏感性明显增高,SDH2缺失导致白念珠菌药物外排能力下降。 结论 SDH2缺失会导致白念珠菌对环境适应性的改变,包括对外界环境压力应答的改变和对唑类抗真菌药物敏感性的增加,以SDH2为靶基因,开发真菌特异性SDH2抑制剂,有望发现与唑类药物协同的新型抗真菌药物。 Abstract:Objective To investigate the role of SDH2 gene in the environmental adaptability of Candida albicans. Methods Wild-type C. albicans strain SC5314, SDH2 gene knockout mutant sdh2 Δ/Δ and reintegrated strain sdh2 Δ /SDH2 were used as experimental objects. Spot assay was conducted to assess the sensitivity of the WT C. albicans strain SC5314, SDH2 gene knockout mutant sdh2 Δ/Δ and reintegrated strain sdh2 Δ /SDH2 to external stress stimulants and antifungal drugs. The effect of SDH2 gene deletion on drug efflux ability of C. albicans was determined by rhodamine 6G efflux assay. Results After SDH2 gene deletion, C. albicans showed slight tolerance to cell wall stress stimulants caffeine, oxidative stress stimulators diamide and menadione. Notably, the sensitivity of SDH2 gene knockout mutant sdh2 Δ/Δ to azole antifungal drugs was significantly increased. The drug efflux capacity of C. albicans was decreased due to the deletion of SDH2 gene. Conclusion SDH2 gene deletion lead to changes in environmental adaptability of C. albicans, including changes in response to external environmental stress and increased sensitivity to azole antifungal drugs. The development of fungal-specific inhibitor targeting SDH2 gene may lead to the discovery of new antifungal drugs which have synergistic effect with azole drugs. -
Key words:
- Candida albicans /
- SDH2 /
- environmental adaptability /
- stress response /
- drug sensitivity
-
近年来,我国药品审评审批制度逐步完善,2005年原国家食品药品监督管理局发布《药品特别审批程序》,对突发公共事件亟需的应急药品可进行特别审批[1]。2016年原国家食品药品监督管理总局发布的《总局关于解决药品注册申请积压实行优先审评审批的意见》[2],首次明确了优先审评审批的范围、程序和工作要求。2020年国家药品监督管理局发布《突破性治疗药物审评工作程序(试行)》等3个文件的公告[3],进一步明确了3种药品加快审批途径。这一系列应急审批程序及加快审评工作规范的发布,对于应急药品供应、创新药物研发具有重要意义:①药品审批效率不断提高,创新药的研发时间明显缩短,适用患者可尽早获得需要的药物治疗;②激励创新药研发机构加大投入并提高创新能力;③在面对诸如新型冠状病毒肺炎(COVID-19)疫情时,开辟绿色通道,相关疫苗及治疗药物迅速上市,在抗击疫情过程中发挥了重要作用。但总体来说,我国的药品应急审批制度起步较晚,实践过程中还在不断摸索,该研究通过分析对比我国与美国、欧盟、日本等国家与地区的药品应急审批制度,探讨进一步优化我国药品应急审批制度的思路和措施。
1. 我国实施药品应急审批的相关制度及应用现状
国家市场监督管理总局2020年1月发布的《药品注册管理办法》[4],将我国药品加快上市注册程序分为突破性治疗、附条件批准、优先审评审批和特别审批程序。该4种程序的适用范围和审批阶段见表1。
表 1 我国药品应急审批程序对比名称 适用范围 申请阶段 突破性治疗药物 在药物临床试验期间,用于防治严重危及生命或严重影响生存质量的疾病且尚无有效防治手段或与现有治疗手段相比具有明显临床优势的创新药或改良新药等 在Ⅰ、Ⅱ期临床试验阶段,通常不晚于Ⅲ期临床试验开展前 附条件批准 治疗严重危及生命且尚无有效治疗手段的疾病的药品,药物临床试验已有数据证实疗效并能预测其临床价值的;公共卫生方面急需的药品,药物临床试验已有数据显示疗效并能预测其临床价值的;应对重大突发公共卫生事件急需的疫苗或者国家卫生健康委员会认定急需的其他疫苗,经评估获益大于风险的 药物临床试验期间 优先审评审批 临床急需的短缺药品、防治重大传染病和罕见病等疾病的创新药和改良型新药;符合儿童生理特征的儿童用药品新品种、剂型和规格;疾病预防、控制急需的疫苗和创新疫苗;纳入突破性治疗药物程序的药品;符合附条件批准的药品 上市许可申请前 特别审批 突发公共卫生事件时,国家药品监督管理局依法决定的应急所需防治药品 提出注册申请前 1.1 突破性治疗
纳入到“突破性治疗”审评通道的药物,药审中心会优先处理有关沟通交流,加强指导并促进药物研发进程;在申报上市环节,该药物可纳入优先审评审批程序,审评时限缩短;上市申请阶段,药审中心会滚动接收其申报资料,并优先安排核查、检验等,这一系列措施可大大缩减新药从研发到上市的时间。2020−2022年间,共有12个药品通过突破性治疗程序上市(表2)。
表 2 2016−2022年我国加快审批途经的注册申请数及获批品种数年份
(年)突破性治疗(件) 附条件批准的
品种数(个)优先审评 特别审批的
注册申请(件)纳入的注册申请 批准的新药上市申请 纳入的注册申请(件) 批准上市品种数(个) 2016 − − − 193 7 − 2017 − − − 230 50 − 2018 − − − 313 83 − 2019 − − − 253 82 − 2020 24 0 6 219 121 59 2021 53 5 38 115 131 81 2022 56 7 31 74 75 51 注:数据来源于NMPA官网。 1.2 附条件批准
“附条件批准”目的在于缩短药物临床试验的时间,使其尽早应用于无法继续等待的危重疾病或公共卫生方面急需的患者。符合附条件批准上市情形的药物,可使用替代终点、中间临床终点或早期临床试验数据来反映药物的有效性,当这些数据能够提示药品的获益大于风险时候,即可申请附条件批准上市。2020−2022年间,共有75个药品通过附条件批准程序上市(表2)。附条件批准可以有效缩短临床研发所需的时间,但也存在一定的风险。因对药品上市时临床安全性等要求的降低,增加了疗效不确定的可能性[5],存在安全有效性不足和资金浪费的风险,对于此类尚无充分证据的药品,平衡满足突发公共卫生事件药品需求和临床安全性可控,科学设计附条件批准方案,是监管部门面临的重要问题。
1.3 优先审评审批
“优先审评审批程序”自2016年发布以来,在实践经验基础上不断优化调整,适用范围更多地向具有明显临床价值、临床急需的药物聚焦,致力于将更多的临床价值显著、临床急需的短缺药品、防治重大传染病、罕见病、儿童用药、纳入突破性治疗程序、符合附条件批准的药品等纳入优先审评程序。同时加速审评时限,药品上市许可申请的审评时限一般为200个工作日,优先审评审批程序的审评时限缩短至130个工作日,其中临床急需境外已上市罕见病用药优先审评审批程序的审评时限为70个工作日。至2022年,共有1 300余件注册申请被纳入该程序,其中获批上市品种数为549个(表2)。
1.4 特别审批程序
“特别审批程序”其核心为出现突发公共卫生事件时,国家药品监督管理部门按照统一指挥、早期介入、快速高效、科学审批的原则,对应急处理所需药品进行特别审批,在申请受理、技术审评、抽样检验、行政审查等环节中优先处置,缩短审批时间。新冠肺炎疫情期间,特别审批程序在新冠病毒疫苗和治疗药物的审批中发挥了重大作用。2020年,共计59件与抗击新冠有关的中药、化学药、生物制品注册申请纳入该程序并完成技术审评,其中建议附条件批准上市1件,建议批准临床试验申请53件,增加适应证的补充申请5件。2021年与2022年分别审结81件及51件纳入特别审批程序的注册申请(均为新冠病毒疫苗和治疗药物),见表2。特别审批程序是一项制度性突破,但随着时间推移,该程序逐渐暴露出实施细节模糊、终止程序缺失等问题[6],不能适应当今国家公共卫生形势的新变化。
我国药品监管部门正在积极推进药品加快上市审评审批制度的改革,并形成初步的监管体制。2023年3月,药审中心总结抗疫应急审批经验,结合已有快速审批制度,制定了《药审中心加快创新药上市许可申请审评工作规范(试行)》[7]。随着改革的发展,更多深层次的问题也会随之暴露。为从根本上解决审评时限长、效率低等问题,药品监管部门应借鉴发达国家的一些加快审评审批政策,并结合中国国情构建一个具有中国特色的药品应急审评审批机制。
2. 国外实施药品应急审批的相关制度及现状
2.1 美国
美国食品药品监督管理局(FDA)建立了4种药品应急审批的途径,包括快速通道(FT)、突破性疗法认证(BT)、优先审评(PR)及加速审批(AA)[8]。① FT:FT可以在药物研发的任何阶段由企业向FDA申请,主要针对在治疗严重的或危及生命的疾病方面具有一定潜力的新药审批。对进入FT的药物,FDA将进行早期介入,以使该药物“少走弯路”,加快研发进程。此外,申请人还可以在早期与FDA沟通,分阶段递交申报资料,不必一次性提交完整的申报资料,而FDA的审批将基于所治疗疾病的严重程度,按风险/效益原则进行评价。②BT:BT主要针对临床试验周期较长的药物,要求申请者提交早期临床试验数据,在药物临床试验阶段助力审批加速。③PR:PR在申请新药上市时提出,FDA会在60 d内做出是否批准优先审评的决定,主要针对与已上市药品比较有显著改进的药品上市申请,并不要求是具有全新分子实体的创新药,但临床疗效必须优于已上市药物。PR的时间为6个月,PR通道并不影响临床试验阶段的周期长短,也不降低审评标准。④AA:AA可使用替代终点、中间临床终点作为许可基础,允许在确切的治疗效果证据未全部收集到之前批准新药上市,主要针对治疗严重的、危及生命的疾病的新药,其批准一般附有条件,即药品具有可观察到的重大短期临床效果,而长期临床疗效则需进一步研究证明。这4种程序之间既存在差异又相互关联,同一个药品申请可适用多种应急审批途经。2018−2022年,FDA年均批准近50个新药,其中约68%的新药使用了一个或多个应急审批途径,具体见表3。
表 3 2018−2022年FDA通过加快审批途经批准的新药情况年份
(年)批准新药总数(个) 加快审批途经批准的新药数量(个) 使用1个或多个加快途经的新药数量及
占批准总数的百分比[个(%)]快速通道 突破性治疗 优先审查 加速审批 2018 59 24 14 43 4 43(72.9) 2019 48 17 13 28 9 29(60.4) 2020 53 17 22 30 12 36(67.9) 2021 50 18 14 34 14 37(74.0) 2022 37 12 13 21 6 24(64.9) 注:数据来源于FDA官网。 此外,美国还建立了紧急使用授权(EUA)制度,即公共卫生处于紧急状态或存在陷入紧急状态的危险时,FDA可对未批准上市的医药产品或已获批准上市医药产品的其他用途进行授权[9]。同时,一旦官方宣布紧急情况结束时,所有基于该声明发布的EUA将不再有效。EUA的发布、更新和终止通知将在《联邦公报》(FR)上公布,并在FDA与美国疾病控制与预防中心(CDC)官网上公告。自2004年EUA制度建立以来,2009年之前FDA只发布过2个EUA。2009−2010年为应对H1N1流感,FDA为22个产品发布EUA,包括药品、诊断试剂和医疗器械。2012年以来,FDA先后对H7N9流感病毒、埃博拉病毒、寨卡病毒、COVID-19等发布多个EUA产品。截至2023年6月,FDA共批准了15个COVID-19治疗药物和4个疫苗的EUA[10]。
2.2 欧盟
欧洲药品审评管理局(EMA)承担欧洲的药品审评审批工作,拥有来自欧盟各国的超过4 000多名专家组成的团队。EMA采取了多种新药应急审批的途径,包括附条件上市许可(CMA)、AA、特殊情况授权(EC)、优先药物审批(PRIME)等。
CMA主要针对未被满足的医疗需求,允许在临床数据不完整的情况下进行早期批准上市,上市后完成确证性临床试验。CMA的有效期为一年,可每年续签。上市许可持有人必须在规定的时间内履行特定义务,包括完成正在进行的或新的研究,或收集额外的数据。一旦上市许可持有人履行了所规定的义务,并且完整的数据证实该药物的益处继续大于其风险,上市许可就可以转换为标准上市许可(不再受特定义务的约束)。对于任何药物,如果新数据显示该药物的益处不再大于其风险,EMA可以暂停或撤销上市许可。2006−2016年间,EMA共有30个附条件上市药物获批,其中11个转为标准授权,2个因为商业原因撤回,其余17个仍处于附条件上市中[11]。
AA可将审批时间从标准程序的210 d缩短至150 d。申请人应证明其申报的医药产品预计具有重大公共卫生利益,特别是从治疗创新的角度来看。另外,申请人应提供有关GMP和GCP方面的信息,以便将常规GCP和批准前的GMP检查纳入加速评估程序。
EC允许患者获得无法根据标准授权批准的药物,在特殊情况下,EMA可能在没有全面数据的情况下授予上市许可。无法获得全面数据的原因在于只有极少数患者患有这种疾病,或是收集有关药物疗效和安全性的完整信息是不道德的。与附条件的上市许可不同,特殊情况授权可能在授权后也无法获得全面的数据。这些药物取得特定的授权后有义务接受EMA的监测约束。
此外,EMA还于2016年3月启动了PRIME计划,该计划目的在于加强对未满足医疗需求的药物,尤其是有潜力带来重大治疗成果的药物开发的支持。PRIME建立在现有的监管框架基础上,通过尽早与药物研发企业合作,为企业提供科学建议和加速评估,并确保患者只参与旨在生成必要数据的必要试验,从而充分利用有限的资源,使药物能够更早地到达患者手中。2016年1月至2021年6月,共有384个药物申请加入PRIME,其中95个被纳入该计划,年平均纳入率为25% [12]。2019−2022年EMA通过AA途经批准的新药情况见表4。
表 4 2019−2022年EMA通过加快审批途经批准的新药年份
(年)批准新药
总数(个)加快审批途经批准的新药数量(个) 附条件
上市许可加速审批 优先药物
审批特殊情况
授权2019 66 8 3 0 1 2020 97 13 6 8 5 2021 92 13 3 6 4 2022 89 9 5 8 5 注:数据来源于EMA官网。 2.3 日本
日本药品与医疗器械管理局(PMDA)负责新药审批,药品应急审批程序包括优先审评、先驱审查认定、附条件审批、再生医学产品有条件和有时限的批准、紧急授权、特例审批等。“优先审评”主要用于具有重大临床价值创新药物及孤儿药的注册审评,2021年,PMDA共批准了144个新药,其中56个通过优先审评,平均审批时间为223 d,同期标准审批时间为300 d。同时,PMDA为了减少不同审查员之间可能存在的人为因素干扰,制定了细致的审查标准化手册,改善了不同机构之间可能存在的信息传递滞后、人员沟通困难等问题,将药品审批时间大幅缩短。
“先驱审查认定”制度自2015年起开始试行,2020年正式提升到法规级别[13]。申请先驱审查的药品需满足治疗方法的突破性与革新性,针对重大严重影响生命质量的疾病或无法根治的疾病等要求。纳入先驱审查认定制度的药品享有优先咨询、加强预评估、优先审查等权力,该制度的审批时限为6个月,比以往减少一半。截至2023年6月,共有200余个药品申请加入先驱审查认定,其中25个药品被成功纳入,该25个药品中的17个已获批上市[14]。其余快速审批制度的对比见表5,通过快速审批,PMDA共批准了9个新冠治疗药,8个新冠疫苗产品(截至2022年末)[15]。
表 5 日本平时与紧急情况下的药品审批制度对比对比项目 平时根据药品性质进行审批 紧急情况下的快速审批 附条件审批 再生医学产品有条件和
有时限的审批特例审批 紧急授权 对象 罕见病用药产品、开创性用药产品或特殊用途用药产品以及其他有特殊医疗需求的用药产品 非同源再生医学及其他产品(细胞/组织产品、基因产
品等)在外国(拥有与日本医药制度同等标准的制度的国家)销售的医药产品和其他产品 所有医药产品 制度宗旨 对医疗需求量大,但很难对足够数量的受试者进行临床试验以验证其疗效和安全性的医药产品给予批准 考虑到再生医学产品的特点(产品质量参差不齐,药理作用表现不一),对那些经少量病例证实安全且假定有效的产品予以批准 为了在紧急情况下防止健康危害的扩散,批准在外国销售的医药产品等 药品和其他产品的安全性已得到确认,其疗效也已得到推定,因此可获得批准,以防止紧急情况下健康危害的扩散 有效性 确认 推定 确认 推定 安全性 确认 确认 确认 确认 2.4 国外相关做法的优势
美国、欧盟、日本的药品优先审评目标定位明确,均以临床需求为目的,旨在加快临床急需药品的审评上市,形成覆盖全流程的多通道、多机制、多模式优先审评体系。制度中具有明确的适用范围、纳入标准、具体申请、受理、审核、审评流程及相关细则。法规体系层次清晰、完整,可操作性强。药审部门制定沟通方案和计划、提前沟通、主动指导。同时,为保证上市药品安全有效,各国针对通过优先审评上市的药品建立了更为严格的上市后监管制度,明确职能定位、监管范围、监管方式、申请人责任和义务,确保监管的科学、规范、有效。美国、日本在推进药品监管领域加快上市注册程序同时,还设置紧急授权使用药品作为应对公共卫生或突发事件的措施。以上这些,都对我国有很好的借鉴意义。
3. 我国药品应急审批制度的思考及启示
3.1 完善药品应急审批制度建设
确定药品需求导向,进一步优化应急审批制度中的药品纳入范围,明确和细化纳入标准。建立符合我国国情的药品优先审评上市后的监管制度,对药品上市后研究、限制性适用、信息公开、撤销上市或转为普通上市,以及相关的强制性手段、法律责任等都应予以明确细化的规定[16]。
建立我国药品EUA制度。我国《中华人民共和国突发事件应对法》对突发事件的预防和应急准备等作出了明确规定,但目前仍缺少关于药品紧急使用授权的法律依据。建议建立我国药品EUA制度,组建由国家卫生健康委员会、药品监管部门及相关部门组成的EUA专业领导工作组协调、管理、实施EUA。应急情况下可考虑对未经药品监管部门批准上市的药品,在具有基础研发数据,可以证明其安全性和有效性的前提下,启动EUA。EUA是在公共健康利益受到严重威胁和药品安全有效评价之间寻找的平衡,遵循“边审批、边使用、边评估、边调整”的原则,可以与加快注册评审同步进行,及时补充调整新药的使用限制。同时,出台EUA实施细则,明确规定实施EUA的条件、发布程序、审评流程和要求、终止或撤销、产品覆盖范围、信息公开、告知义务、责任保护等[17]。
3.2 增强政府职能和优化人员配置
目前的药品应急审批程序主要从促进申请人与药审中心沟通、调整上市标准这两方面来提高新药注册的效率。我国虽设置了药品审评专家咨询委员会,但人员配置数量较欧盟、日本等较少,在药品审评过程中需要召开会议解决问题,或是申请过程中存在争议主动征询意见时提供相应的技术指导与决策建议,发挥作用有限。应积极推动审评审批人才培养和队伍建设,扩大药审专家职能,实现人员配置的优化和审评通过率的提升。
药品的上市审批过程中,申请方与监管部门、检验机构之间存在信息交流滞后的问题,应尽快推广和加强药审中心受理人员及项目管理人员的提前介入,争取受理问题在申报前得到解决,降低企业的创新风险,同时防止审评资源的浪费与人力、物力的消耗。
3.3 加强快速审批药品上市后监管
美国、欧盟、日本药品优先审评制度中,药审部门侧重沟通指导,并重视上市后药品的安全性与有效性,建立了严格的上市后监管制度。现阶段,我国加快上市注册程序对新药的开发促进作用较为明显,通过快速审批程序注册上市的药品逐年增多,为避免相关药物上市后出现无期望的临床疗效或存在安全性问题,急需加强快速审批药品上市后的监管工作,保证审批药物的安全有效。新版《药品注册管理办法》引入药品“加快上市注册程序”的同时也明确设立了上市退出机制。为实现此类特殊审评通道药品上市前、后安全监管工作的顺利衔接,可借鉴EMA的经验,细化特殊审评药品准入条件,将上市前风险管理体系的建立作为关键审评因素,上市后设置额外监测、风险沟通等安全监管制度进行有效衔接,从而实现对用药风险的持续监测和动态管理。
4. 结语
随着公众对药品上市的效率以及对突发事件应急药品供应的需求提升,我国药监部门也尽力在保障药品安全有效的前提下,出台一系列政策以尽量缩短紧急必需用药的审评审批时间。在应对新冠疫情过程中,药品应急审批工作已取得了诸多进展。然而,对比国外药品快速审批程序,我国目前仍处于发展阶段,因此需认真总结既往的工作经验,并在实践过程中不断完善。同时借鉴一些国外先进模式和监管方式,进一步改善我国药品应急审批制度体系和流程,提升药品审批效率,提高应对突发事件的药品保障能力。
-
[1] PFALLER M A, DIEKEMA D J. Epidemiology of invasive mycoses in North America[J]. Crit Rev Microbiol,2010,36(1):1-53. doi: 10.3109/10408410903241444 [2] ENOCH D A, YANG H N, ALIYU S H, et al. The changing epidemiology of invasive fungal infections[J]. Methods Mol Biol,2017,1508:17-65. [3] BROWN G D, DENNING D W, GOW N A, et al. Hidden killers: human fungal infections[J]. Sci Transl Med,2012,4(165):165rv13. [4] ENE I V, ADYA A K, WEHMEIER S, et al. Host carbon sources modulate cell wall architecture, drug resistance and virulence in a fungal pathogen[J]. Cell Microbiol,2012,14(9):1319-1335. doi: 10.1111/j.1462-5822.2012.01813.x [5] SUN N, FONZI W, CHEN H, et al. Azole susceptibility and transcriptome profiling in Candida albicans mitochondrial electron transport chain complex I mutants[J]. Antimicrob Agents Chemother,2013,57(1):532-542. doi: 10.1128/AAC.01520-12 [6] THOMAS E, ROMAN E, CLAYPOOL S, et al. Mitochondria influence CDR1 efflux pump activity, Hog1-mediated oxidative stress pathway, iron homeostasis, and ergosterol levels in Candida albicans[J]. Antimicrob Agents Chemother,2013,57(11):5580-5599. doi: 10.1128/AAC.00889-13 [7] BI S, LV Q Z, WANG T T, et al. SDH2 is involved in proper hypha formation and virulence in Candida albicans[J]. Future Microbiol,2018,13:1141-1156. doi: 10.2217/fmb-2018-0033 [8] OYEDOTUN K S, LEMIRE B D. The quaternary structure of the Saccharomyces cerevisiae succinate dehydrogenase. Homology modeling, cofactor docking, and molecular dynamics simulation studies[J]. J Biol Chem,2004,279(10):9424-9431. doi: 10.1074/jbc.M311876200 [9] HANS S, FATIMA Z, HAMEED S. Insights into the modulatory effect of magnesium on efflux mechanisms of Candida albicans reveal inhibition of ATP binding cassette multidrug transporters and dysfunctional mitochondria[J]. Biometals,2021,34(2):329-339. doi: 10.1007/s10534-020-00282-w -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4025
- HTML全文浏览量: 1333
- PDF下载量: 32
- 被引次数: 0
