

 下载:
下载:
 下载:
下载:

-
急性髓系白血病(AML)是原始和幼稚细胞在骨髓或外周血中恶性克隆、异常增殖的一种恶性血液系统肿瘤疾病,其高复发率、高病死率成为临床治疗的关键与难点[1-3]。在AML的成人患者中,约30%发生了FMS 样酪氨酸激酶(FLT3)突变,使得FLT3成为治疗AML的重要靶点之一[4-5]。吉瑞替尼作为第二代FLT3抑制剂,可同时靶向FLT3的基因内部串联重复(ITD)和酪氨酸激酶结构域(TKD)两个位点,抗肿瘤活性更广,不良反应发生概率更低,是国内外指南中治疗FLT3突变的复发、难治性AML患者的推荐用药[6-7]。吉瑞替尼常见的不良反应有:丙氨酸氨基转移酶升高(25.4%)、天冬氨酸氨基转氨酶升高(24.5%)、贫血(20.1%);其他具有临床意义的严重不良反应包括:QTc间期延长(0.9%)、可逆性后脑部病综合征(0.3%) [8-9]。尽管不良反应发生概率相对较低,但严重不良反应仍使吉瑞替尼在临床应用上受限。该研究报道了临床药师参与1例吉瑞替尼在治疗过程中导致获得性QTc间期延长的药学监护,探讨临床药师在肿瘤药物治疗过程中的重要作用,同时为吉瑞替尼的临床应用提供参考。
-
患者,女,76岁,身高155 cm,体质量44 kg,体表面积1.36 m2。2022年12月,感染新型冠状病毒后痊愈,但偶感乏力,活动后症状加重,遂于当地医院就诊,血常规提示:白细胞 72.97×109 /L; 血红蛋白 49.2 g/L; 血小板 101×109 /L;骨髓涂片示幼稚细胞占61%,医师对症予以2 U O型悬浮红细胞改善贫血,建议行骨穿及MICM检查并转诊专科治疗。后为进一步治疗,于2023年2月15日由急诊以“贫血待查,乏力”收治入火箭军特色医学中心(本院)。否认其他传染病史和慢性病史,否认药物、食物过敏史。
-
入院后完善各项常规检查。血常规:白细胞 102.73×109 /L; 血红蛋白 49.2 g/L; 血小板 85×109 /L;生化:血钾3.32 mmol/L;骨髓涂片示幼稚细胞占75%,根据骨髓免疫分型及细胞形态学检查结果确诊为急性髓系白血病,FAB分型为急性单核细胞白血病(AML-M5)。医师于2月20日行维奈克拉(口服,第1天100 mg,第2天200 mg,第3~28天400 mg)+阿扎胞苷(皮下注射,第1~7天75 mg/m2)方案治疗。3月5日基因检测结果回报,患者FLT3基因突变阳性,遂于3月6日起,加用富马酸吉瑞替尼片(口服,120 mg d1~28)。药师建议,可在患者吉瑞替尼开始用药前、首次用药第8天和第15天进行心电图检查。根据心电图检测回报,患者在用药前、用药第8、15天的QTc数值分别为: 402、475、487 ms,因患者QTc间期延长超过60 ms,药师建议立即停用可疑药品吉瑞替尼。停药第3天复查心电图,结果显示QTc数值为424 ms,恢复至正常范围内。心电图监测结果如图1所示。
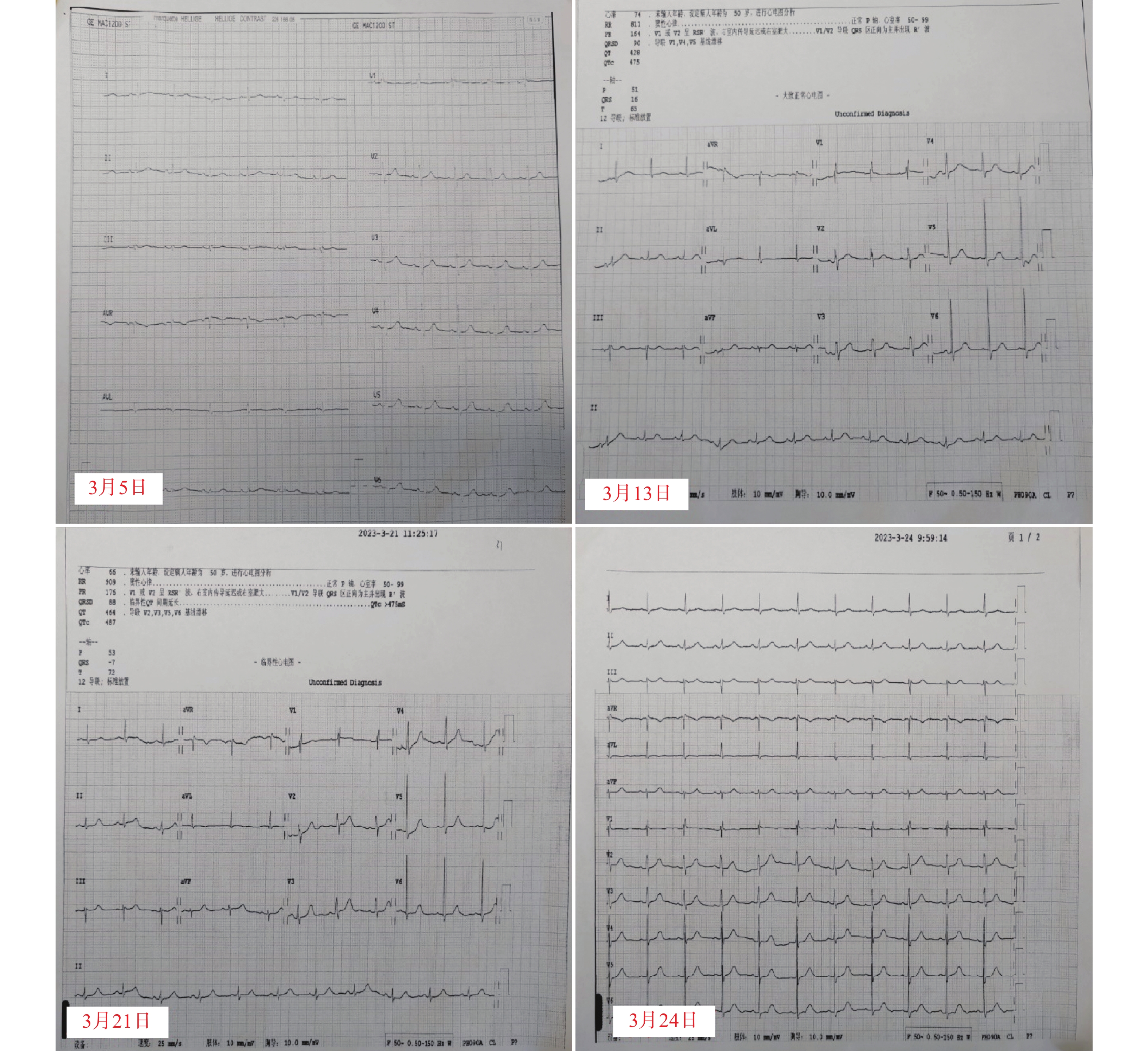
图 1 患者吉瑞替尼用药前1 d、用药后第8天、第15天以及停药后第3天心电图变化情况
-
国内尚未对QTc间期延长做统一定论,一般将QTc数值>440 ms作为QTc间期延长的界值,但也存在部分正常人QTc数值超过这个正常值范围[10]。美国心脏协会、美国心脏病学学会认为男性QTc 470 ms,女性480 ms为正常值。但不论男女,QTc>500 ms或QTc变化数值较基线值变化超过60 ms都属于明显异常,且在美国卫生及公共服务部发布的常见不良事件评价标准(CTCAE v5.0)中,将QTc≥501 ms或比基线期>60 ms定义为QTc间期延长的3级不良反应事件。本例患者用药期间QTC数值变化近90 ms,QTc间期明显延长,及时查明可疑药品,予以对症处理十分必要。
患者自述既往无心脏病史,吉瑞替尼用药前心电图结果正常,可排除患者自身疾病影响。根据国家药品监督管理局、药品不良反应中心对于判定不良反应事件相关性分析原则[11]:①患者吉瑞替尼用药前QTc结果正常,3月6日用药后,QTc数值不断上升,因此不良反应发生与用药存在时间关系;②通过查阅说明书和相关文献可知,吉瑞替尼具有引起患者QTc间期延长的风险;③患者停药后,QTc数值逐渐恢复正常;④本周期未再使用吉瑞替尼;⑤患者同期应用的其他治疗药物,例如:维奈克拉、伏立康唑、美罗培南均为长期用药,且吉瑞替尼停药后这些药物仍在使用。综上所述,可判断患者QTc间期延长的不良反应很可能是由吉瑞替尼引起的。
-
QTc间期通常是指心室除极化和复极化的总耗时,也就是自QRS波起点至T波终点所需的时间[12]。长QTc间期综合征(LQTS),是一种心室复极时程延长、不均一性增大的疾病。心电图上表现为 QTc间期延长、T波和(或)U波异常、早搏后的代偿间歇,且心率减慢时可发生尖端扭转型室性心动过速(TdP),严重时可诱发猝死。临床表现为心动过缓、晕厥、搐搦,包括先天性和获取性两类[10]。该患者既往无心脏疾病史及家族遗传史,且入院时QTc数值正常,可排除先天性LQTS。获取性LQTS与药物、心脏疾病、代谢异常等因素相关,其中常见的可引起LQTS药物有:抗心律失常药、抗生素、抗精神失常药、抗肿瘤药等[13-14]。
吉瑞替尼致QTc间期延长机制研究较少,通常认为药源性LQTS的发生与抑制电压依赖性K+通道电流有关,主要为延迟整流K+通道(IK)。其中,快速激活(Ikr)者的α亚基由hERG基因编码,是整个动作电位复极时相的主要电流。若Ikr通道受阻,则可造成心脏动作电位时程中QTc间期延长,心室复极化延迟,进而诱发TdP [15-16]。患者入院时生化结果示:血钾3.32 mmol/L,低于正常值范围。钾离子对于维持心肌细胞自律性、传导性和兴奋性具有重要意义。低血钾时,Ikr通道外向复极电流减弱,动作电位复极延迟,多表现为心脏自律性增强及传导减慢,是诱发LQTS的重要因素。又因为吉瑞替尼在体内主要经CYP3A酶代谢,是CYP酶系中易受合用药物影响的亚型,而患者化疗期间,出现Ⅳ度骨髓抑制(白细胞0.48×109 /L,血红蛋白82 g/L,血小板17×109 /L,中性粒细胞绝对值0.03×109 /L),间断发热,医师给予注射用美罗培南1 g 1/8 h 静滴+伏立康唑200 mg 1/12 h 静滴(用药时间3月13日至3月26日)对症治疗。伏立康唑属于强效CYP3A/P-gp抑制剂,可有效抑制CYP3A酶活性,使得患者体内吉瑞替尼代谢减慢,血液浓度可增加1.5倍左右。因此,不良反应发生率随之上升。本例患者使用吉瑞替尼后出现QTc间期延长的3级不良反应,可能与患者血钾水平低、联合使用CYP3A/P-gp抑制剂引起药物暴露水平升高有关。
-
一旦发生药源性LQTS,首先要判断可疑药品,立即停用明确或可能进一步诱发尖端扭转型室性心动过速的药物,并进行连续的QTc间期监测,积极对症治疗。临床药师可通过详细询问患者既往病史,仔细审查用药医嘱,判断引起不良反应的可能药品,通过关联性评价确定引发不良反应的可能性大小,分析用药医嘱中是否存在合并用药的相互作用增加患者QTc间期延长风险。同时评估是否存在其他诱发或加重患者LQTS发生风险的高危因素。本例患者联用伏立康唑会增加吉瑞替尼血药浓度,故临床药师建议吉瑞替尼用药过程中,避免与强效CYP3A/P-gp抑制剂合用,同时及时补钾,改变低血钾状态,防止TdP发生。该患者吉瑞替尼停药后,密切监护其心电图变化情况,3 d后QTc数值恢复至正常范围内(424 ms)。临床药师建议可酌情恢复用药,以80 mg的减量剂量继续用药,恢复用药后患者未再出现心电图明显异常。目前患者规律服用该药,病情控制平稳。建议在后续2个周期治疗开始前进行心电图检查,同时提醒患者若出现心慌、胸闷、晕厥要立即到医院就诊。若不能自行恢复,可静脉注射硫酸镁治疗[10,17],同时在后续2个周期治疗开始前进行心电图检查。
-
国内外对于吉瑞替尼引起QTc间期延长的报道十分罕见,QTc间期延长的不良反应往往不易被发现,最终对患者产生严重危害。临床药师通过了解已知或可能的存在诱发LQTS或TdP风险相关的药物,个体化评估药源性QTc间期延长风险及临床获益情况,判断合并用药是否存在相互作用情况,及其他并发的高危因素,并通过适当减少用药剂量、及时纠正电解质紊乱等方法,来降低不良反应发生概率,提高用药安全性。
Participation of clinical pharmacists in QTc interval prolongation induced by gilteritinib
-
摘要:
目的 探讨临床药师在1例急性髓系白血病患者应用吉瑞替尼引起QTc间期延长的病例中的作用,为此类患者的药物治疗和监护提供参考。 方法 临床药师及时发现1例急性髓系白血病患者心电图异常情况,通过分析患者基础疾病、诊疗过程、治疗用药及其潜在相互作用参与临床诊疗。 结果 临床药师怀疑QTc间期延长很可能是吉瑞替尼引起的不良反应,建议立即停用该药,复查心电图。医师采纳此建议,及时停止可疑药品吉瑞替尼药物治疗,3 d后复查心电图,患者QTc数值恢复至正常范围内。 结论 临床药师参与临床诊疗过程,可为患者提供更优质的药学服务。 Abstract:Objective To explore the role of clinical pharmacists involved in the case of a patient with acute myeloid leukemia whose QTc interval prolongation was induced by gilteritinib, and to provide reference for drug treatment and monitoring of those patients. Methods The abnormal electrocardiogram (ECG) of a patient with acute myeloid leukemia was found in time by clinical pharmacists, who participated in clinical diagnosis and treatment by analyzing the patient’s underlying diseases, diagnosis and treatment process, therapeutic drugs and their potential interactions. Results Clinical pharmacists suspected that the prolonged QTc interval was likely to be an adverse reaction caused by gilteritinib, and recommended immediate discontinuation of the drug and re-examination of the electrocardiogram.The physician took the suggestion to stop the suspected drug therapy with gilteritinib promptly, and ECG was rechecked 3 d later, and the QTc value returned to the normal range. Conclusion Clinical pharmacists participating in clinical diagnosis and treatment could provide better pharmaceutical care for patients. -
Key words:
- gilteritinib /
- QTc interval prolongation /
- adverse drug reaction /
- clinical pharmacists
-
光动力治疗(PDT)基于光辐照聚集光敏剂的肿瘤组织,由光敏剂诱发光动力反应形成单线态氧(1O2)等活性氧(ROS),通过对肿瘤细胞和肿瘤血管的直接杀伤及激活机体系统免疫反应等多种机制发挥抗肿瘤作用[1-3]。二氢卟吩及菌绿素类光敏剂是PDT新药研究的热点[4-8]。其中,已获批上市的代表药物有他拉泊芬(talaporfin)和帕利泊芬(padeliporfin)等[9, 10]。
光敏剂作为结构非特异性药物,存在缺乏肿瘤靶向性摄入和明确的作用药靶等缺陷。此外,PDT受制于局部治疗,对浸润较深的肿瘤组织,及已发生转移的肿瘤疗效有限。目前,PDT和化疗联用是克服上述缺陷,提高PDT疗效最为普遍和有效的策略之一。研究表明,抗代谢化疗药物氟尿嘧啶(5-Fu)与PDT联用具有协同抗肿瘤作用[11-13]。据此,我们设想利用在肿瘤微环境下能响应性断裂的连接基团(linker)将光敏剂与化疗药物偶联,希望实现二者在肿瘤组织的靶向释放,从而发挥其PDT和化疗协同抗肿瘤作用。酰腙键是酸敏感化学键,常被用来连接载体,以药物制备智能药物载体。这种药物载体到达肿瘤细胞的内涵体或溶酶体中时,会发生酸性水解将药物有效释放出来。因此,本文针对肿瘤微环境呈弱酸性的特点,采用药物化学最经典的前药设计策略,以脱镁叶绿素a(Phephorbide a)粗提物经酸碱降解制得的二氢卟吩e6(3)[14]为先导光敏剂,通过其152-羧基与抗肿瘤药物5-Fu以酸敏感酰腙键连接,设计合成pH响应型光化疗协同抗肿瘤光敏剂二氢卟吩e6-偕氟尿嘧啶(1),并考察其体外PDT抗肿瘤活性和pH响应性5-Fu释放,及其对黑色素瘤B16-F10和肝癌HepG2细胞的光动力抗癌活性及其作用机制,以期获得高效、低毒的PDT治癌药物候选药物,合成路线见图1。
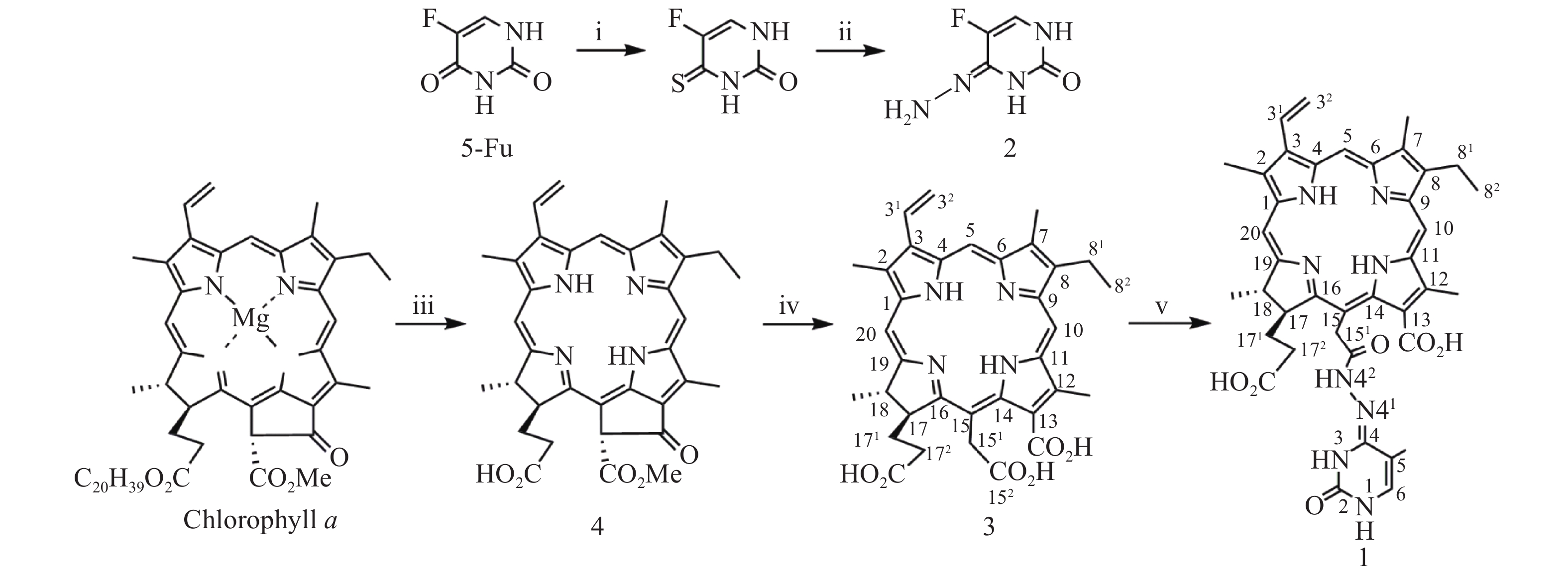 图 1 二氢卟吩e6 -偕氟尿嘧啶光敏剂(1)的合成路线试剂和反应条件:(i)五硫化二磷,吡啶,回流12 h;(ii)水合肼,甲醇,室温2 h;(iii)浓盐酸,乙醚,4 ℃ 30 min;(iv)25%氢氧化钾甲醇液,回流30 min;(v)a. EDC·HCl,N,N-二甲基甲酰胺,室温8 h;b.二异丙基乙胺,2, N,N-二甲基甲酰胺,室温12 h。
图 1 二氢卟吩e6 -偕氟尿嘧啶光敏剂(1)的合成路线试剂和反应条件:(i)五硫化二磷,吡啶,回流12 h;(ii)水合肼,甲醇,室温2 h;(iii)浓盐酸,乙醚,4 ℃ 30 min;(iv)25%氢氧化钾甲醇液,回流30 min;(v)a. EDC·HCl,N,N-二甲基甲酰胺,室温8 h;b.二异丙基乙胺,2, N,N-二甲基甲酰胺,室温12 h。1. 化学合成
1.1 仪器与试剂
用Bruker MSL-600型核磁共振仪测定1H NMR,CD3OD为溶剂;用API-3000 LC-MS型电喷雾质谱仪测定质谱(ESI-MS);用岛津UV-160型紫外分光光度计测定UV吸收谱;用日立F-7000荧光分光光度计测定荧光发射谱;用Shimazu LC-20AD HPLC仪测定化合物1的相对纯度及其5-Fu的体外释放。色谱柱型号为Waters Xterra C18柱,流动相:乙腈-0.3%乙酸水溶液(80 : 20);流速:1.0 ml/min;检测波长:400 nm(化合物1的相对纯度)或254 nm(5-Fu释放);柱温:30 ℃;进样量:20 μl。柱色谱分离用TELEDYNE ISCO的快速制备色谱Combi Flash@Rf+仪,硅胶H作为固定相。PDT抗癌活性测试使用BWT半导体激光仪(北京凯普林,波长为660 nm);用流式细胞仪(BD Accuri C6,美国)(激发波长:488 nm,发射波长:525 nm)检测受试肿瘤细胞样品的ROS水平、细胞凋亡率和细胞周期阻滞。
二氢卟吩e6(3)按照文献[14]的方法制备;其它实验用材料和化学试剂均为市售商品。
1.2 42-N-(二氢卟吩 e6-152-酰基)-5-氟尿嘧啶-4-腙(1)的合成
取氟尿嘧啶(0.2 g,1.563 mmol)溶于无水吡啶(10 ml),加入五硫化二磷(0.298 g,1.563 mmol),加热回流12 h。反应完毕,减压回收溶剂,残物加乙酸乙酯溶解(100 ml),用0.1 mol/L HCl洗涤(50 ml×2),无水Na2SO4干燥,减压除去溶剂得4-硫代-5-氟尿嘧啶粗品。上述4-硫代-5-氟尿嘧啶粗品加甲醇(10 ml)溶解,于0 ℃下滴加N2H4·H2O(0.316 g,6.252 mmol),室温继续搅拌2 h。反应完毕,减压抽滤,P2O5真空干燥得固体化合物5-氟尿嘧啶-4-腙(2)中间体,直接用于下步反应。取二氢卟吩e6(0.1 g,0.168 mmol)溶于无水DMF(10 ml),加1-乙基-(3-二甲氨基丙基)碳二亚胺盐酸盐(EDC·HCl)(0.035 g,0.183 mmol),室温搅拌反应6 h后再加入中间体2(0.031 g,0.218 mmol),继续搅拌36 h。反应完毕,反应液加入10倍体积量乙酸乙酯,饱和NaCl水溶液洗涤(50 ml×3),无水Na2SO4干燥,减压回收溶剂所得固体经快速制备色谱梯度洗脱分离纯化(流动相为二氯甲烷/甲醇/甲酸=15∶1∶0.1~8∶1∶0.1)得黑色固体1纯品0.048 g,产率39.6%。UV-vis λmax (MeOH, nm) (ε, M−1cm−1):660 (3.15×104), 510 (0.82×104), 402 (8.13×104)。1H-NMR (600 MHz, CD3OD, δ, ppm): 9.79 (s, 1H, 10-CH), 9.73 (s, 1H, 5-CH), 9.07 (s, 1H, 20-CH), 8.19 (dd, J = 18.0, 12.0 Hz, 1H, 31-CH), 7.29 (s, 1H, 5-Fu的6-CH), 6.38 (d, J = 18.0 Hz, 1H, 32-CHB), 6.15 (d, J = 12.0 Hz, 1H, 32-CHA), 5.35 (s, 2H, 151-CH2), 4.65 (m, 2H, 17-CH和18-CH), 3.84 (q, J = 7.5 Hz, 2H, 81-CH2), 3.63 (s, 3H, 12-CH3), 3.53 (s, 3H, 2-CH3), 3.30 (s, 3H, 7-CH3), 2.3~2.0 (m,4H , 171-CH2 和172-CH2), 1.76 (m, 6H¸ 18-CH3和82-CH3)。MS (ESI+) m/z: 723.63 (M+H)+ (100%)。元素分析(C38H39N8O6F,%)计算值:C 63.16, H 5.40, N 15.48;实测值:C 63.34, H 5.38, N 15.43。HPLC测定纯度:95.2%。
2. 体外光理化性质和光生物活性
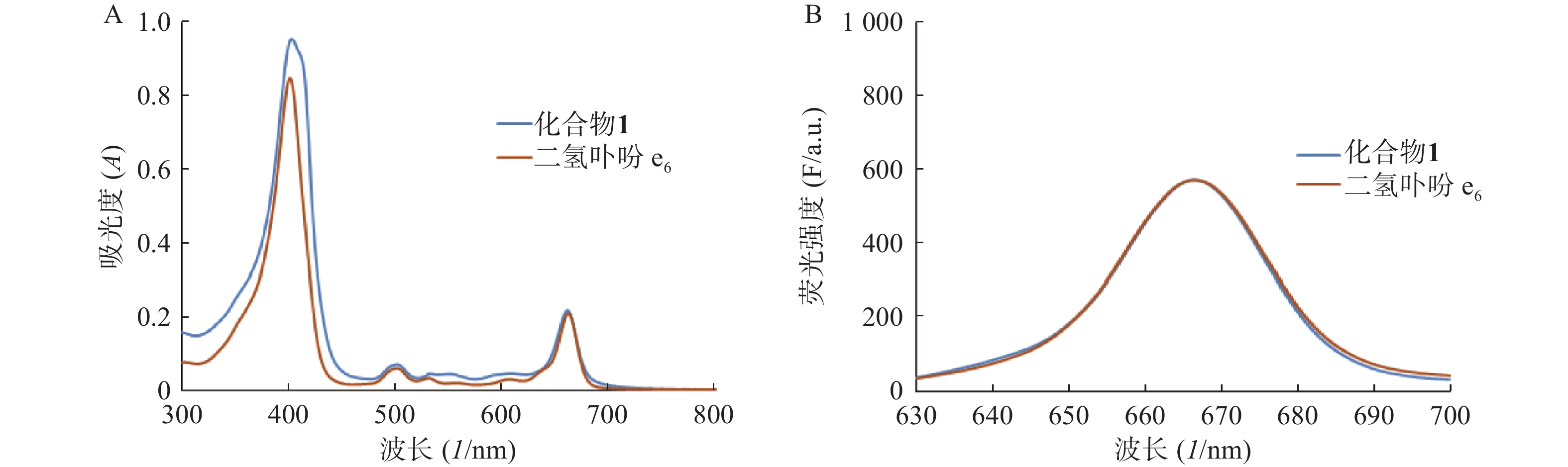
2.1 化合物1的紫外吸收谱和荧光发射谱
分别测定目标化合物1及其先导化合物二氢卟吩e6(3)的甲醇溶液(10 μmol/L)在300~800 nm处的紫外吸收谱和激发波长为400 nm的荧光发射光谱,结果见图2。
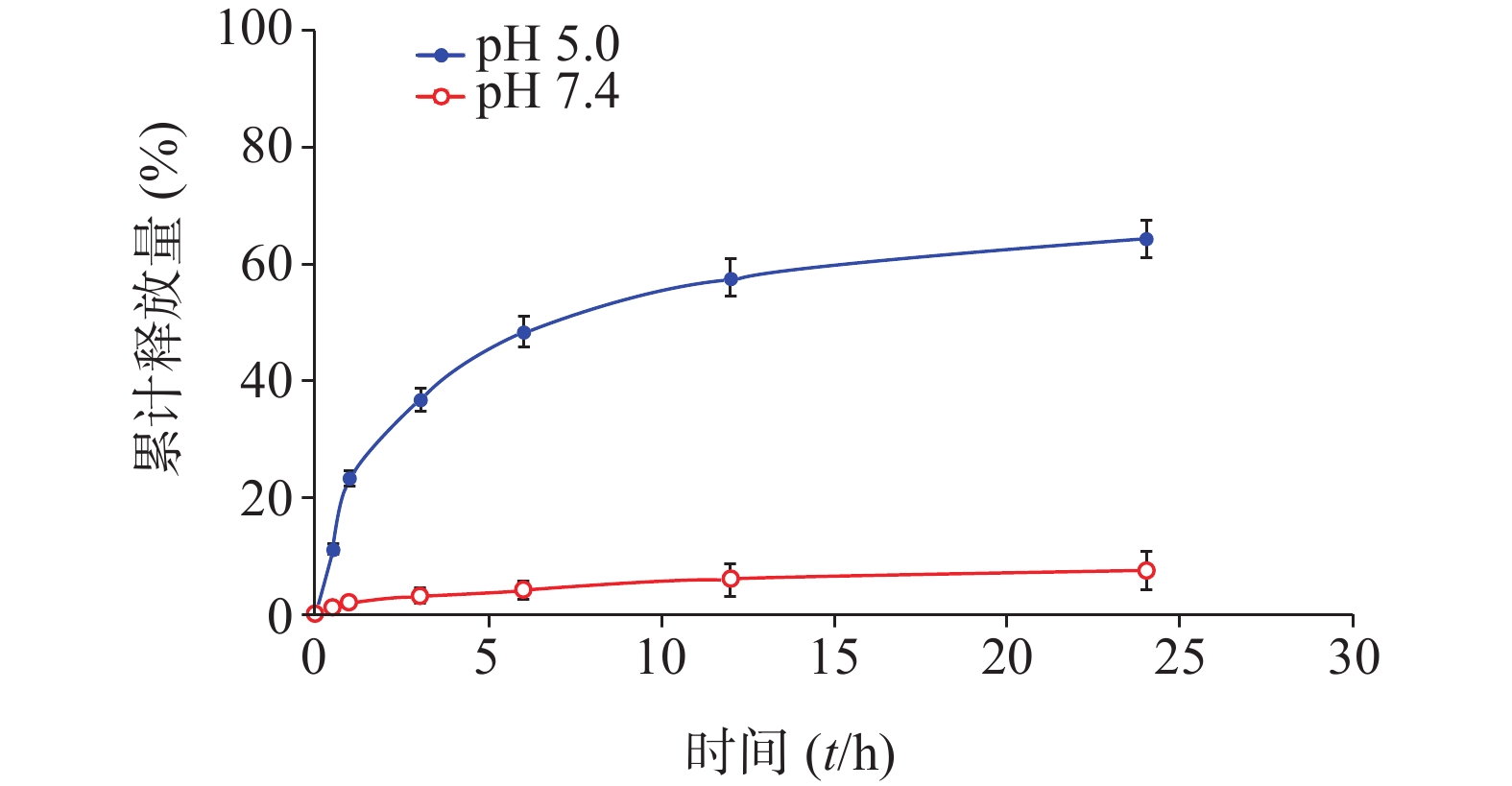
2.2 化合物1的体外pH响应性5-Fu释放
分别配制浓度为50 μmol/L的化合物1的HOAc-NaOAc缓冲液(pH 5.0)和PBS溶液(10 ml),并于0.5、1.0、3.0、6.0、12、24 h时分别取样(500 μl)。其中,HOAc-NaOAc缓冲液(pH 5.0)组取样液用0.1 mol/L氢氧化钠水溶液迅速调节pH值至7.4。每份取样液加PBS稀释至原溶液1/3浓度,微孔滤膜(孔径0.22 μm)过滤,HPLC进样检测;实验重复3次。根据5-Fu的HPLC峰面积-浓度标准曲线分析计算,绘制目标化合物1于弱酸(pH 5.0)中的5-Fu体外释放量-时间曲线,结果见图3。
2.3 化合物1的体外光动力抗癌活性
2.3.1 细胞孵育
2.3.2 细胞暗毒性测试
参照文献[6-8]的方法,每孔5×103个B16-F10细胞或HepG2细胞悬液(100 μl)接种于96孔板上,加入等体积上述细胞培养液孵育24 h;更换含不同浓度待测物的培养液(DMSO浓度小于1%,100 μl),继续避光孵育48 h;再更换含10%(V/V)CCK-8(Beyotime,中国)的RPMI 1640基础培养基(100 μl),继续培养1.5 h,然后用Varioskan Flash全波长酶标仪(Thermo)于波长450 nm处测定每孔的吸光度值,计算各浓度对应的细胞存活率,并拟合得到待测物的肿瘤细胞半数抑制浓度即IC50值。
2.3.3 细胞光毒性测试
每孔5×103个B16-F10细胞或HepG2细胞悬液(100 μl)接种于96孔板上,加入等体积细胞培养液孵育24 h;更换含不同浓度待测物的细胞培养液(DMSO浓度小于1%,100 μl),继续避光孵育24 h;再更换新鲜培养液(100 μl),以波长为660 nm的激光辐照受试细胞样品(光照剂量为10 J/cm2),继续孵育24 h。最后按“2.3.2”项下CCK-8法测定各待测物的肿瘤细胞IC50值。
2.3.4 实验结果
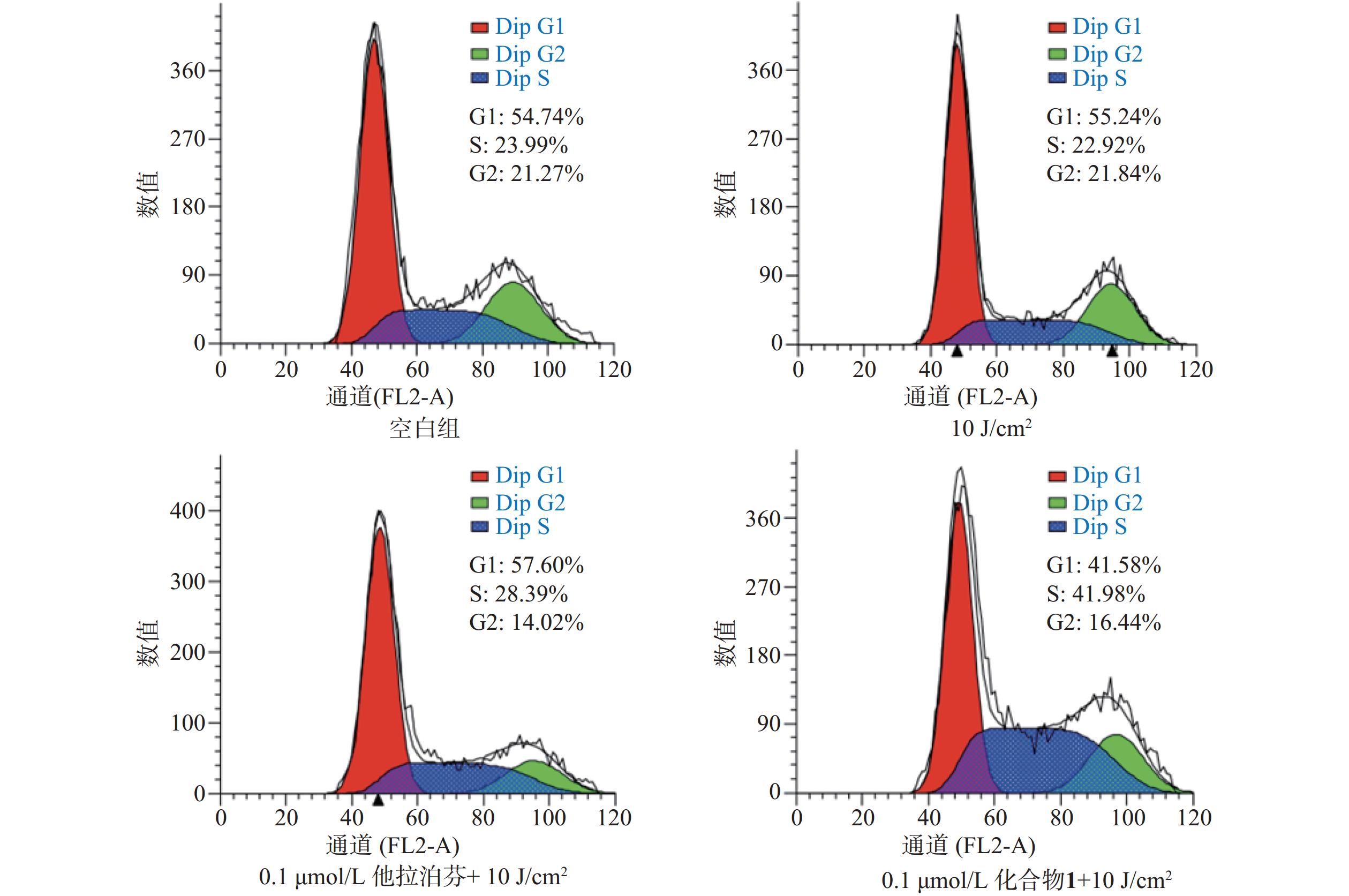
以临床光敏药物他拉泊芬为阳性对照,化合物1及其先导化合物3对肿瘤细胞株的体外PDT抗癌活性结果见表1。
表 1 目标化合物1的体外光动力抗癌活性(IC50,μmol/L)化合物 B16-F10细胞 暗毒/光毒比 HepG2细胞 暗毒/光毒比 暗毒性 光毒性 暗毒性 光毒性 化合物 1 46.84±8.46*, ΔΔΔ 0.73±0.16**, ΔΔΔ 64.2 50.80±6.45**, #, ΔΔΔ 0.90±0.22**, ΔΔΔ 56.4 二氢卟吩e6 69.72±4.69 3.36±0.59 20.8 70.38±10.9 2.75±0.41 25.6 他拉泊芬 254.8±18.8 11.31±3.88 22.5 176.4±28.4 15.47±5.07 11.4 5-Fu 35.80±6.68 NTa − 39.16±2.7 NTa − NTa:未测定;*P < 0.05,**P < 0.01,与二氢卟吩 e6组比较;#P < 0.05,与5-Fu组比较;ΔΔΔP < 0.001,与他拉泊芬组比较。 2.4 化合物1介导的PDT对肿瘤细胞内ROS水平的影响
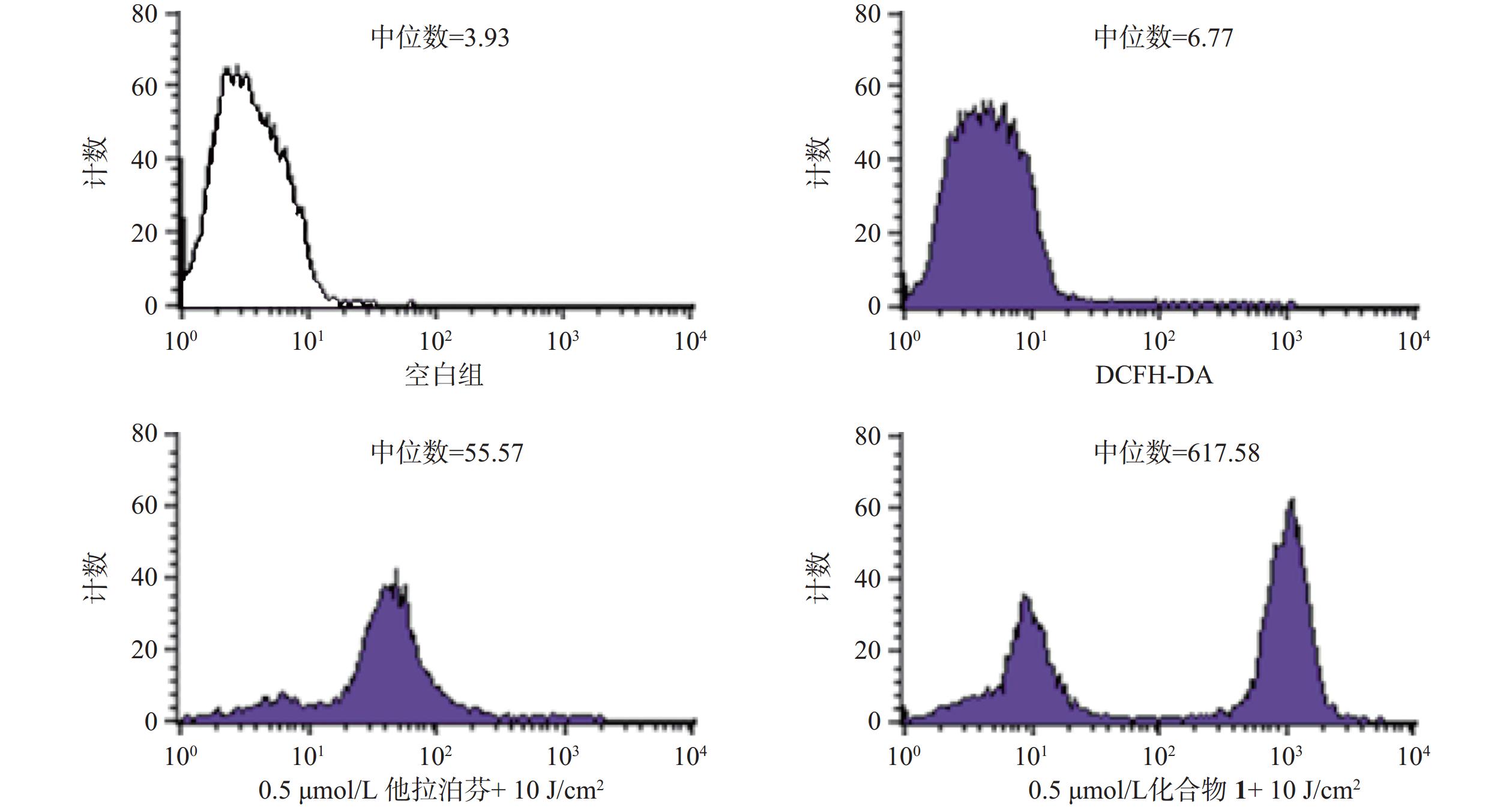
操作步骤如下:a. 每孔3 × 105个B16-F10细胞悬液(2 ml)接种6孔板上,按“2.3.1”项条件避光孵育24 h;b. 分别更换含一定浓度化合物1或他拉泊芬的新鲜培养液(DMSO浓度小于1%,2 ml),继续避光孵育24 h;c. 加入10 mmol/L DCFH-DAROS荧光检测探针(Beyotime,1.5 μl),吹打混匀,继续避光孵育20 min;d. PBS洗涤3次,再加新鲜培养液(2 ml),以660 nm波长的激光辐照(光剂量10 J/cm2)细胞样品,继续避光孵育20 min;e. 收集每孔细胞样品,用流式细胞仪检测各孔细胞ROS水平,结果见图4。
2.5 化合物1介导的PDT对肿瘤细胞凋亡的影响
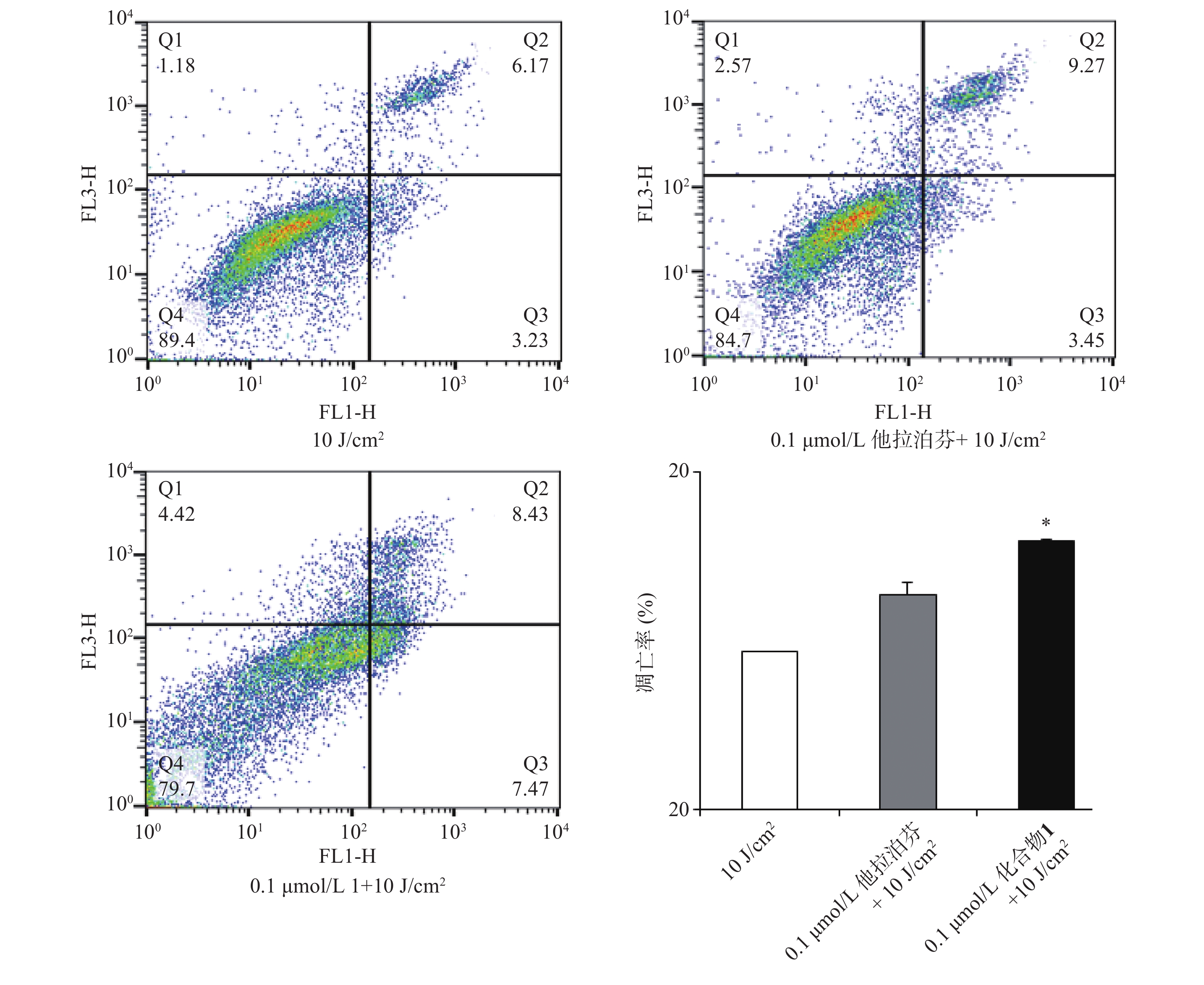
按“2.4”项下操作方法,仅从步骤c开始,更换新鲜培养液(2 ml),用660 nm波长的激光辐照(光剂量10 J/cm2)细胞样品,继续避光孵育20 min;d. 以1 500 r/min离心(5 min)细胞样品,PBS洗涤,再以1 000 r/min离心(5 min)后获取细胞样品;e. 按Annexin V-FITC细胞凋亡检测试剂盒(Beyotime)操作流程操作,结果见图5。
2.6 化合物1介导的PDT对肿瘤细胞周期的阻滞作用
按“2.5”项下操作方法,仅在e步骤中,换以细胞周期阻滞检测试剂盒(Beyotime)的操作流程,每份细胞样品中分别加入染色缓冲液(300 µl)、RNase A(6 µl)和碘化丙啶染色液(15 µl),轻轻混匀,避光孵育20 min后,用流式细胞仪进行细胞周期阻滞检测,结果见图6。
3. 结果与讨论
按文献[14]方法制得的二氢卟吩e6(3)为先导化合物,经1-乙基-(3-二甲氨基丙基)碳二亚胺盐酸盐(EDC·HCl)于无水DMF中催化分子内脱水缩合制得二氢卟吩e6-131,152-酸酐活泼中间体[15],然后直接与中间体2发生酰化反应成功合成得到了光化疗双模抗肿瘤光敏剂二氢卟吩e6-偕氟尿嘧啶(1),反应收率达39.6%,其结构经UV、ESI-MS、1H NMR及元素分析确证。
化合物1在甲醇中最大紫外吸收波长和荧光发射波长(激发波长:400 nm)分别为660 nm和670 nm,与先导物3相一致,表明先导物3以酰腙键偶联5-Fu后,并没有改变其作为光敏剂特有的紫外吸收和荧光发射光谱等光物理特性。此外,化合物1在弱酸(pH 5.0)条件下,能有效释放5-Fu,24 h内累积释放率可达60.3%;但在pH 7.4的条件下较为稳定,24 h内5-Fu累积释放率仅为5%。
体外PDT抗癌活性测试结果显示,化合物1对B16-F10和HepG2细胞株的光毒活性和暗毒/光毒比(治疗指数)均显著优于先导物二氢卟吩e6(3)(P<0.005)和他拉卟吩(P<0.001),其IC50值分别达0.73 μmol/L和0.90 μmol/L。
体外PDT抗癌机制研究提示,化合物1介导的PDT能显著提升B16-F10细胞内ROS水平和诱导B16-F10细胞凋亡,并阻滞肿瘤细胞周期于S期。
总之,二氢卟吩e6-偕氟尿嘧啶(1)具有PDT抗癌活性强、治疗指数(暗毒/光毒比)高且可在肿瘤弱酸环境中有效释放5-Fu等优点,从而实现“单分子”光化疗协同抗肿瘤作用,值得进一步开发研究。
-
[1] LIU H T. Emerging agents and regimens for AML[J]. J Hematol Oncol, 2021, 14(1):1-20. doi: 10.1186/s13045-020-01025-7 [2] 李慧, 庄海峰. 急性髓系白血病伴FLT3-ITD突变研究进展[J]. 中国实用内科杂志, 2022, 42(4):340-344. [3] 朱昆, 于倩, 郭义明, 等. 自体造血干细胞移植急性髓系白血病患者的药学监护与实践1例[J]. 中南药学, 2019, 17(10):1754-1758. [4] ZHONG Y E, QIU R Z, SUN S L, et al. Small-molecule fms-like tyrosine kinase 3 inhibitors: an attractive and efficient method for the treatment of acute myeloid leukemia[J]. J Med Chem, 2020, 63(21):12403-12428. doi: 10.1021/acs.jmedchem.0c00696 [5] LARROSA-GARCIA M, BAER M R. FLT3 inhibitors in acute myeloid leukemia: current status and future directions[J]. Mol Cancer Ther, 2017, 16(6):991-1001. doi: 10.1158/1535-7163.MCT-16-0876 [6] 袁伟, 张世忠, 主鸿鹄. FLT3抑制剂治疗急性髓系白血病患者研究进展[J]. 浙江大学学报(医学版), 2022, 51(4):507-514. [7] LEE L Y, HERNANDEZ D, RAJKHOWA T, et al. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor[J]. Blood, 2017, 129(2):257-260. doi: 10.1182/blood-2016-10-745133 [8] PERL A E, MARTINELLI G, CORTES J E, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML[J]. N Engl J Med, 2019, 381(18):1728-1740. doi: 10.1056/NEJMoa1902688 [9] NUMAN Y Z, ABDEL RAHMAN Z, GRENET J, et al. Gilteritinib clinical activity in relapsed/refractory FLT3 mutated acute myeloid leukemia previously treated with FLT3 inhibitors[J]. Am J Hematol, 2022, 97(3):322-328. doi: 10.1002/ajh.26447 [10] 中华医学会心血管病学分会心律失常学组, 中国心脏起搏与心电生理杂志编辑委员会, 中华心血管病杂志编辑委员会. 获得性长QT间期综合征的防治建议[J]. 中国心脏起搏与心电生理杂志, 2010, 24(6):471-479. [11] 杨华, 魏晶, 王嘉仡, 等. 药品不良反应/事件报告评价方法研究[J]. 中国药物警戒, 2009, 6(10):581-584. doi: 10.3969/j.issn.1672-8629.2009.10.002 [12] 周翠翠, 王鸿. QT间期延长的临床研究进展[J]. 医学综述, 2011, 17(15):2313-2315. [13] TISDALE J E. Drug-induced QT interval prolongation and torsades de pointes: role of the pharmacist in risk assessment, prevention and management[J]. Can Pharm J, 2016, 149(3):139-152. doi: 10.1177/1715163516641136 [14] 戎佩佩, 陈敏, 刘虹, 等. 克唑替尼致获得性长QT间期综合征的病例报告并文献复习[J]. 实用药物与临床, 2020, 23(8):717-720. [15] MORISSETTE P, HREICHE R, TURGEON J. Drug-induced long QT syndrome and torsade de pointes[J]. Can J Cardiol, 2005, 21(10):857-864. [16] 王骏, 严铭玉, 王鸣和. 药源性QT延长综合征的研究进展[J]. 世界临床药物, 2007, 28(3):152-156. [17] 冉拓耀, 李超. 哌柏西利胶囊致QT间期延长1例分析[J]. 中国药物警戒, 2023, 20(2):215-218. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5708
- HTML全文浏览量: 1480
- PDF下载量: 22
- 被引次数: 0
