


 下载:
下载:
 下载:
下载:

-
嘧啶类化疗药物在肿瘤治疗中的地位越来越重要,其代表药物5-氟尿嘧啶及其口服前药卡培他滨更受到了广泛关注,体内二氢尿嘧啶脱氢酶(DPD)是此类药物代谢的限速酶之一[1],前瞻性评价DPD的总体活性有利于提高药物疗效及减少患者的毒副反应,对临床具有重要意义。内源性物质尿嘧啶(U)是体内DPD的天然底物,在此酶的催化下生成二氢尿嘧啶(UH2),并最终通过尿液排出体外。测定血浆中U和UH2的含量,并通过(UH2)/(U)比值计算,可从代谢物的角度评价DPD的活性[2]。临床上常用评价DPD酶活性的方法是测定患者的基因表型,DPD的编码基因DPYD序列中包含了多达7 600个多态位点,使得DPD酶的活性在人群中是高度可变的[3]。不同的突变位点及不同位点的组合给临床检测带来了极大的困难。到目前为止,也只有DPYD*2A的多态性被用于临床实践,用来筛选出5-氟尿嘧啶代谢严重不良的患者,避免严重的毒副反应[4]。单一应用基因的多态性来评价DPD酶的活性在临床上存在一定的困难,基因的多态性并不能直接同下游的酶的活性联系起来,两者并没有完全对等的关系。基因需通过转录、翻译和蛋白的修饰之后才能发挥作用。基因多态联合下游代谢物的含量测定更能准确的评价DPD酶的活性[5]。目前常应用液相色谱-串联质谱联用法对人血浆或干燥唾液中U和UH2浓度进行检测[6-11],但所报道的方法均有一些复杂或难以重现。本研究成功的建立了一种灵敏、高效、准确、重现性好,且能同时测定人血浆中U和UH2浓度的UHPLC-MS/MS方法,为体内DPD总体活性提供更客观有效的评价途径。
-
1290-6460A超高效液相色谱-串联质谱仪,包含G4220A二元泵、G4226A自动进样器、G1316C柱温箱、MassHunter数据处理工作站(美国Agilent);调速涡旋混合器(美国Labnet);SK7200H超声仪(上海科导超声仪器有限公司);BSA124S-CW分析天平(德国赛多利斯);5810R型低温高速离心机、移液器(德国Eppendorf公司)。
-
尿嘧啶、二氢尿嘧啶和氯尿嘧啶(内标)对照品(纯度>99%,大连美仑生物有限公司);乙酸铵(美国赛默飞世尔科技);甲醇、乙腈、乙酸乙酯、异丙醇(色谱纯,德国默克公司);屈臣氏蒸馏水(广州屈臣氏食品饮料有限公司);牛血清白蛋白(BSA)(上海博光生物科技有限公司);生理盐水(长征医院药学部自制)。
-
色谱柱为Agilent poroshell 120 SB-Aq-柱(2.1 mm×100 mm,2.7 μm),流动相为5 mmol/L乙酸铵水溶液(A)和乙腈(B),流速为0.3 ml/min,梯度洗脱:0~0.3 min,100% A;0.3~1.0 min,100%~10% A;1.0~2.5 min,10% A;柱温为30 ℃,洗脱时间2.5 min,进样量5 μl。
-
采用ESI离子源,多重反应监测(MRM)进行一/二级质谱分析,用于定量分析的检测离子为:U[M+H]+ m/z 113.0→40.1,检测模式为正离子模式;UH2[M+H]+ m/z 115.0→55.1,检测模式为正离子模式;氯尿嘧啶(IS)[M-H]- m/z 145.0→42.1,检测模式为负离子模式。雾化温度为300 ℃,雾化气压力为40 psi,干燥气流速为10 L/min,鞘气温度300 ℃,鞘气流速12 L/min,解离电压为4 000 V。
-
选取8名血浆样品指标正常的成年人,于当日清晨8时空腹状态下静脉采血3 ml, EDTA-3K管抗凝,离心后分离上层血浆, 于−80 ℃冰箱冻存。
-
取100 μl样本,加10 μg/ml氯尿嘧啶(IS)10 μl,加乙酸乙酯3 ml,涡旋5 min,1710×g离心10 min,取上层有机相2.7 ml,45 ℃氮气挥干仪挥干,用10%甲醇溶液100 μl复溶,涡旋1 min,取上清液进样分析。
-
用含有3 %牛血清白蛋白作为空白基质代替血浆配置标准曲线样品。取100 μg/ml的尿嘧啶和二氢尿嘧啶各100 μl,加800 μl水,制成10 μg/ml标准混合液,置于−20 ℃备用。取10 μg/ml标准混合液适量,用3 %牛血清白蛋白稀释制成10、20、50、100、200、500、1000、1500 ng/ml系列浓度样品, 然后按照上述“1.6”项下样品的处理方法配制。
-
U和UH2的出峰时间以及峰型良好,代替血浆经过前处理后,对待测组分的测定没有干扰,内标对分析物的测定也没有干扰,且能很好分离,结果见图1。
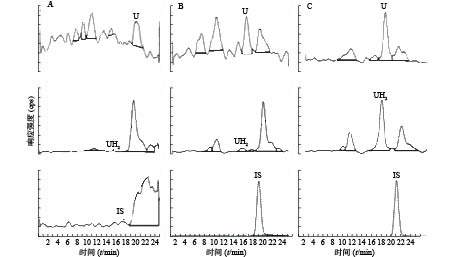
图 1 尿嘧啶和二氢尿嘧啶及内标质谱多反应监测色谱图
-
U和UH2的线性范围是10.0~1 500.0 ng/ml,以空白BAS中U和UH2的浓度为横坐标(X),U和UH2与内标化合物氯尿嘧啶的峰面积比为纵坐标(Y),进行最小二乘法加权(权重系数为1/χ2),U和UH2的线性回归方程分别是Y=0.27X+0.0022、Y=0.58X+0.0380,r均>0.990,表明线性关系良好。
-
取定量下限、低、中、高标准添加血浆样本按照前处理方法进行处理,每个浓度样品平行制备5份进行分析,连续3 d重复操作,根据当天的标准曲线计算当天实测样本浓度,计算样本在低、中和高浓度下的日内、日间精密度和准确度,结果显示,精密度和准确度的偏差均在15%左右。准确度相对偏差在20%范围内时,最低定量下限精密度偏差不大于20%,结果见表1。
表 1 尿嘧啶和二氢尿嘧啶的精密度 (n=5)
分析物 标示浓度 (ng/ml) 日内 日间 测定浓度 (ng/ml) 精密度(CV%) 准确性(RE%) 测定浓度 (ng/ml) 精密度(CV%) 准确性(RE%) 尿嘧啶 10 10.2±0.38 3.74 2.58 10.12±0.78 7.70 1.18 20 20.63±1.21 5.87 3.15 19.97±1.35 6.74 −0.16 500 529.73±4.64 0.88 5.95 484.32±35.72 7.37 −3.12 1000 1093.33±25.10 2.30 9.33 1098.25±25.16 2.29 9.82 二氢尿嘧啶 10 10.32±0.71 6.86 3.18 10.28±0.65 6.37 2.77 20 19.98±2.19 10.95 −0.12 19.86±1.85 9.31 −0.72 500 517.51±10.69 2.07 3.50 515.66±10.36 2.01 3.13 1000 1079.83±17.91 1.66 7.98 1080.11±24.50 2.27 8.01 -
取低、高2个浓度的样本进行基质效应和提取回收率考察,结果显示,U、UH2及内标氯尿嘧啶的基质效应和提取回收率良好,结果均较稳定,结果见表2。
表 2 尿嘧啶和二氢尿嘧啶的基质效应和提取回收率
分析物 标识浓度(ng/ml) 基质效应 提取回收率 平均基质效应 CV(%) 平均回收率 CV(%) 尿嘧啶 1000 101.00 6.15 94.98 9.01 20 99.99 3.63 100.01 7.64 二氢尿嘧啶 1000 85.72 2.07 106.47 1.58 20 93.58 4.53 99.54 9.77 -
考察低、高2个浓度的血浆样品经历3次冷冻与解冻循环的稳定性、血浆样品在室温(25 ºC)放置6 h后经样品处理后稳定性和血浆样品经样品处理后室温放置24 h的稳定性,结果显示,3次冻融、6 h室温(25 ºC)条件下和24 h放置自动进样器中的稳定性均符合要求,结果见表3。
表 3 样品的稳定性(RE%)
分析物 冻融3次 室温放置6 h 置自动进样器24 h 低 高 低 高 低 高 尿嘧啶 100.71 98.34 93.49 106.60 108.20 107.99 二氢尿嘧啶 92.67 92.64 93.61 107.26 106.97 107.15 -
应用本研究所建立的方法,对8名健康成人的血浆样本测定分析,在样本实测过程中, 同时插入已知浓度的随行质控样本(QC样本), 随时监控样本测定的准确度。U和 UH2 浓度测定结果见图2。
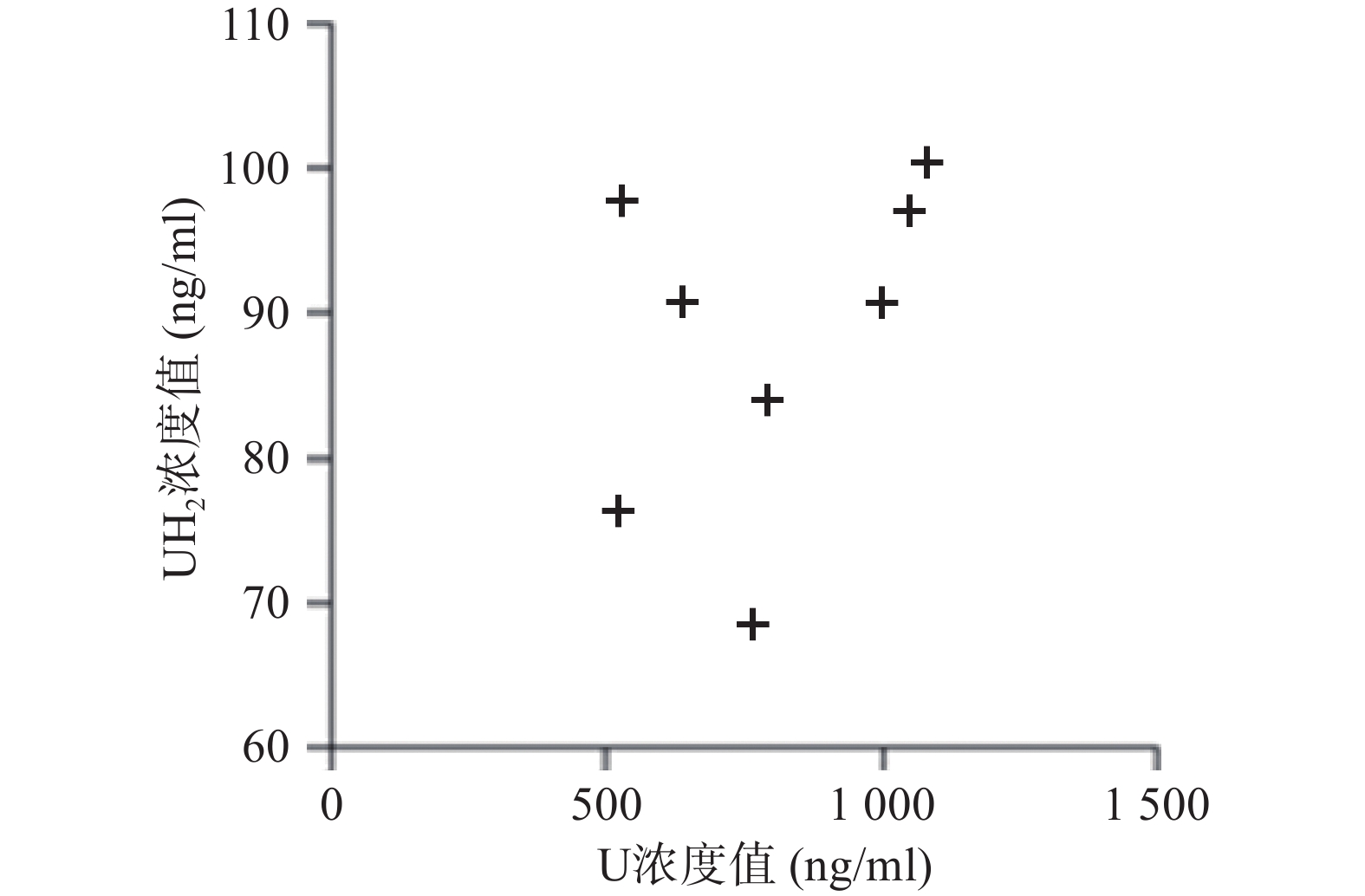
图 2 实测8名健康成人内源性U和UH2浓度值
-
U和UH2是人体内常见的两种物质,且同核酸的代谢密切相关,由于U和UH2均为人体内源性物质,故不能采用人源的基质进行方法学的开发及验证,通过查阅资料,选择了不含U和UH2的3%牛血清蛋白作为基质进行方法学的开发[12]。也有文献报道采用去除U和UH2的人源血浆基质进行方法学实验[9],但基质的来源较珍贵,不适合方法的普及,所以选用3%牛血清蛋白作为替代基质。
-
U因其特殊的化学性质,在大部分的色谱柱上均没有保留。U和UH2的LogP值分别为−0.707和−0.840,有较强的亲水性,决定了其不保留的性质。在定量方法的开发过程中,先后采用了Agilent Zorbax SB-C18色谱柱,Agilent Zorbax Eclipse-C18色谱柱,Waters Atlantis T3色谱柱,Waters Xselect色谱柱,Waters Xbridge等色谱柱来进行条件摸索,但上述色谱柱对U和UH2均没有保留。最后,采用Agilent Zorbax SB-Aq对U和UH2进行定量分析,该色谱柱对强极性的化合物有较好的保留效果,同时,兼容100%的起始流动相也对保留产生了良好的结果。
-
本研究还分别考察了几种常见的处理方法,包括甲醇和乙腈的蛋白沉淀、Waters Oasis HLB萃取板的固相萃取以及乙酸乙酯,甲基叔丁基醚,二氯甲烷/三氯甲烷,环己烷进行的液液萃取,结果发现乙酸乙酯对U和UH2的萃取效果较好,同时,还分别考察了5%、10%、20%、30%、40%、50%的异丙醇、乙酸乙酯溶液对U和UH2的萃取效果,结果发现,单纯的乙酸乙酯对待测化合物具有较好的提取效率。提取回收率均高于90%,且RSD<10%。另外对3%牛血清蛋白的基质效应进行了考察,结果发现平均基质效应在85%~101%之间,RSD<7%,说明该前处理方法对于基质的清除较为彻底,测定结果稳定,没有明显的基质干扰。
本研究虽重在方法开发,收集的样本数量较少,但从测定的U和UH2浓度分布来看DPD对内源性U的代谢存在个体差异,建议临床应用5-氟尿嘧啶及其卡培他滨筛查DPD总体活性[12-13],后续可进一步扩大样本数量进行深入研究。
-
本实验建立了一种快速,稳定,高灵敏度的UHPLC-MS/MS方法,可用于测定人体内源性物质U和UH2的含量,从代谢物的角度评价DPD酶的活性,从而协助临床医生制定化疗药物5-氟尿嘧啶及其口服前药卡培他滨合理的用量,以较低的毒副反应获得最大的临床疗效。
UHPLC-MS/MS determination of uracil and dihydrouracil in human plasma
-
摘要:
目的 建立同时测定人血浆中尿嘧啶(U)和二氢尿嘧啶(UH2)含量的超高效液相色谱串联质谱(UHPLC-MS/MS)方法。 方法 在Agilent 6460A串联质谱仪上采用正离子检测模式,以氯尿嘧啶为内标,3%牛血清蛋白为代血浆基质,样本经乙酸乙酯液液萃取后在Agilent poroshell 120 SB-Aq (2.1 mm×100 mm,2.7 μm)色谱柱上采用梯度洗脱进行色谱分离。流动相为5 mmol/L醋酸铵水溶液和乙腈溶液;流速0.3 ml/min;柱温为30 ℃;进样量为5 μl。 结果 尿嘧啶和二氢尿嘧啶的线性范围为10.0~1500.0 ng/ml,线性关系良好,其相关系数r>0.990,日内与日间精密度偏差均<15%。 结论 该方法操作简单、选择性好,可用于测定人血浆中尿嘧啶和二氢尿嘧啶的含量。 -
关键词:
- 超高效液相串联质谱法 /
- 尿嘧啶 /
- 二氢尿嘧啶 /
- 含量测定
Abstract:Objective To establish an UHPLC-MS/MS method for the determination of uracil (U) and dihydrouracil (UH2) in human plasma. Methods A positive ion detection mode was adopted on the Agilent 6460A mass spectrometer. Chlorouracil was used as the internal standard. 3% bovine serum albumin was used as surrogate plasma matrix. The pretreatment of plasma sample was completed based on liquid-liquid extraction with ethyl acetate. The chromatographic separation was achieved on an Agilent Poroshell 120 SB-Aq (2.1 mm×100 mm, 2.7 μm) column with gradient elution. The mobile phase was 5 mmol/L ammonium acetate aqueous solution and acetonitrile solution. The flow rate was 0.3 ml/min. The column temperature was 30°C. The injection volume was 5 μl. Results The linear range of uracil and dihydrouracil was 10.0-1500.0 ng/ml. Both of uracil and dihydrouracil had good linear relationship with correlation coefficient (r)>0.990. Both of inter- and intra-day precision was <15%. Conclusion The established method is simple, selective, and suitable for the determination of U and UH2 in human plasma. -
Key words:
- UHPLC-MS/MS /
- uracil /
- dihydrouracil /
- content determination
-
甲巯咪唑(MMI)为硫脲类抗甲状腺药物(ATD),是治疗甲状腺功能亢进症的一线药物,其常见的不良反应为过敏性皮肤反应,一般较轻微,罕见的不良反应有血液系统异常(如全血细胞减少)和肝损伤等,若未及时治疗可危及生命。全血细胞减少是指患者未接受过放、化疗,至少连续2次外周血三系细胞数量均低于正常值,即WBC<4.0×109/L(ANC<1.5×109/L)、RBC<3.5(3.0)×1012/L或Hb<110(100)g/L、PLT<100×109/L[1]。据报道,ATD致全血细胞减少的发生率在日本约为0.01%[2],在我国约为0.04%[3],同时合并肝损伤就更为少见。笔者对1例甲巯咪唑致全血细胞减少及肝损伤患者进行病例分析,为治疗该类患者提供用药参考。
1. 病例资料
患者女,30岁,54 kg,因“发热、咽痛、乏力3 d”于2019年10月6日入院。患者6个月前无明显诱因出现怕热多汗、多食易饥、易怒、心悸、失眠症状,7月25日查甲状腺功能:FT3 31.55 pmol/L,TT3 7.38 nmol/L,FT4 85.15 pmol/L,TT4 260.5 nmol/L,TSH<0.005 mIU/L,甲状腺球蛋白抗体(TgAb) 267 IU/ml,甲状腺过氧化物酶抗体(TPOAb) 72.4 IU/ml;肝功能、血常规正常;甲状腺摄碘率:3 h 46.1%,6 h 67.7%,24 h 71.2%;诊断为甲状腺功能亢进症,予甲巯咪唑片10 mg/次,3次/d。9月2日复查甲功:TSH 0.0014 mIU/L,FT3 7.17 pmol/L,FT4 22.51 pmol/L,Anti-TSHR 7.57 IU/L;血常规正常;肝功:ALT 95 IU/L,AST 53 IU/L;予复方甘草酸苷片(含甘草酸苷25 mg)1片/次,3次/d保肝治疗。10月3日患者出现发热、咽痛伴乏力,最高体温40 ℃,自行服用对乙酰氨基酚片0.75 g/次,2次/d。10月5日患者病情无好转,于本院急诊科查血常规:WBC 0.56×109/L,NEUT 0.031×109/L,Hb 94 g/L,PCT 16.24 ng/ml,立即停用甲巯咪唑,予莫西沙星、头孢哌酮舒巴坦、重组人粒细胞刺激因子等治疗1 d,复查血常规:WBC 0.64×109/L,NEUT 0.009×109/L,Hb 97 g/L,为进一步治疗收治入院。患者无心、肝、血液系统疾病史,无药物过敏史,无低碘区居住史。
入院查体:T 40.4 ℃,P 106次/min,R 20次/min,BP 133/68 mmHg;皮肤及巩膜轻度黄染;咽部黏膜充血,扁桃体Ⅱ度肿大、脓性分泌物附着;甲状腺Ⅰ度肿大、质软、无压痛、未扪及结节;右下肢散在黄豆大小皮肤破溃。
入院诊断:甲状腺功能亢进症,中性粒细胞缺乏,化脓性扁桃体炎。
2. 住院期间主要临床信息及药物治疗经过
该患者在本院住院治疗期间的主要临床信息及药物治疗经过详见图1。
3. 讨论
3.1 甲巯咪唑致全血细胞减少及肝损伤的关联性评价
疾病方面,甲亢和严重感染性疾病均可致全血细胞减少。患者出现典型甲亢症状约3个月后开始口服MMI治疗,用药前血常规正常,服药后FT3、FT4降至正常,可排除甲亢导致的全血细胞减少。患者初诊时严重中性粒细胞缺乏、轻度贫血,入院第5天PCT、hsCRP下降明显,仍发展为三系细胞减少,当感染治愈后中性粒细胞未恢复至正常值,可排除严重感染性疾病导致的全血细胞减少。药物方面,无复方甘草酸苷片各组分致全血细胞减少的报道,虽有甘草合剂致血小板减少的个案,但二者关联性不明确[4]。日本一项50 385例的回顾性研究发现,MMI致全血细胞减少的中位时间为41 d(32~97 d),累计剂量为1 200~2 109 mg,但发病机制尚不明确,可能与ATD致中性粒细胞缺乏的机制重叠,当严重的粒细胞缺乏不及时干预可发展为全血细胞减少[2]。患者服用MMI 71 d,累计剂量为2 130 mg,根据Naranjo评估量表患者得分情况如下:该ADR先前有结论性报告(1分)、该ADR是在使用MMI后发生(2分)、存在客观证据证实该ADR与MMI有关(1分),总分4分,故患者全血细胞减少可能与MMI相关。
药物性肝损伤为排他性诊断,患者无肝病史、嗜酒史,经辅助检查可排除甲亢、病毒性肝炎、自身免疫性肝炎、脂肪肝、肝脏占位及胆囊结石导致的肝损伤,故考虑药物因素可能性大。MMI致肝损伤大多发生在用药12周内[5],主要为胆汁淤积型,其次为肝细胞损伤型和混合型。本例肝损伤首先表现为AST和ALT轻度升高,虽服用复方甘草酸苷片仍出现黄疸,TBIL>5ULN,R值=1.74,为胆汁淤积型重度肝损伤[6], 根据RUCAM量表患者得分情况如下:首次服用MMI 39 d后出现肝脏生化学检查异常(2分)、排查其他原因(2分)、MMI说明书中有肝毒性报告(2分),总分6分,故患者肝损伤很可能与MMI相关。
3.2 甲巯咪唑的停药指征
甲亢患者ATD疗程一般为12~18个月,但当ANC≤0.5×109/L[7](或ANC<1.5×109/L[8]),或转氨酶>3 ULN或持续升高,或出现黄疸时应停药。患者入院时NEUT 0.009×109/L,皮肤及巩膜可见黄染,因此需立即停用MMI。由于ATD致粒细胞缺乏可能在再次服药时出现,且ATD之间有交叉反应,不宜换用另一种药物,后续可采用放射性131I或外科手术治疗。
3.3 全血细胞减少伴发热的药物治疗
3.3.1 抗感染治疗
患者10月6日NEUT 0.01×109/L,危险度分层为高危,宜采取降阶梯抗感染的策略,初始方案须覆盖铜绿假单胞菌等严重G-菌[9]。患者使用美罗培南4 d,体温波动于39.5 ℃,扁桃体I度肿大,PCT 0.31 ng/ml,评估抗感染效果不佳。由于患者右下肢皮肤破溃未愈合,不排除该处为感染灶之一,因此,临床药师建议联用万古霉素加强抗金黄色葡萄球菌等G+菌力度,于用药48 h后监测谷浓度以确保万古霉素达有效治疗浓度(10~15 mg/L)。患者经美罗培南联合万古霉素抗感染5 d后,体温下降至36.8 ℃,生命体征平稳,选用头孢哌酮舒巴坦行降阶梯治疗。
3.3.2 升白细胞治疗
临床上通常使用粒细胞-巨噬细胞集落刺激因子(GM-CSF)和粒细胞集落刺激因子(G-CSF)以降低化疗药物引起的粒细胞缺乏者的感染风险,但二者未被批准用于非化疗药物导致的粒细胞缺乏症。GM-CSF用于ATD诱导的粒细胞减少缺少文献报道,且可引起血小板下降。多数作者主张在严重粒细胞缺乏或预后不佳的重症患者中使用G-CSF,以帮助其度过危险期[3, 10]。一项Meta分析表明,G-CSF可有效缩短亚洲人群ATD致粒细胞缺乏的恢复时间[WMD=−3.16 d(95%CI:−4.58~−1.74,P=0.000)][11]。因骨髓中成熟中性粒细胞约2.5×1012个,而原始粒细胞分化为成熟中性粒细胞需7~14 d,故使用G-CSF后,中性粒细胞绝对值(ANC)曲线呈双峰形。首先,G-CSF促进骨髓中成熟粒细胞向外周血释放形成第1峰,由于新的成熟粒细胞未生成,此时不宜停药;其次,G-CSF刺激骨髓粒系造血祖细胞加速增殖、分化、成熟和释放,使ANC降至最低点后再次逐渐上升形成第2个峰[12]。根据CTCAE5.0标准,患者为中性粒细胞减少4级,使用G-CSF 150 μg/d 4 d后,ANC曲线第1个高峰不明显,可能与药物剂量不足或骨髓长时间被抑制有关。G-CSF升高ANC呈剂量依赖性,一般给药剂量为300 μg/d或5 μg/(kg·d),重症患者可根据临床效果增加剂量[13-14],在G-CSF治疗无效时使用小剂量泼尼松可有效升高ANC水平。由于糖皮质激素可抑制免疫反应,患者当前感染严重,因此,临床药师建议将G-CSF的用量增至300 μg/d,同时警惕肌肉、骨骼疼痛等ADR,糖皮质激素仅在G-CSF治疗无效后且在足量抗菌药物的前提下慎重使用[15]。ANC<0.1×109/L是公认的预后不佳的因素之一,可作为G-CSF的停药指征[16]。2015版《临床用药须知》指出,严重感染伴粒细胞减少者,ANC≥1.0×109/L时停用G-CSF。本例患者的ANC经第一个高峰后升至1.593×109/L,此时感染已控制,予以停用G-CSF,合理把握了停药时机。
4. 小结
全血细胞减少合并肝损伤是ATD罕见的严重不良反应,早诊断、早治疗则预后良好,否则可能继发严重感染从而威胁生命。因此,如何防范并及早发现上述不良反应,需临床药师做好用药教育:①建议患者监测血常规,在治疗初期前3个月每周1次,维持治疗期间每月1次,当WBC<4.0×109/L但ANC>1.5×109/L时,通常不用停药,可服用维生素B4等升白细胞药物。②提醒患者服药期间若出现咽喉疼痛、口腔炎、发热等症状,应立即就诊,并告知医生正在服用ATD。③建议患者在治疗初期的前3个月,每月监测1次肝功能,若出现厌食、上腹部疼痛、黄疸等症状时,应立即就诊。
-
表 1 尿嘧啶和二氢尿嘧啶的精密度 (n=5)
分析物 标示浓度 (ng/ml) 日内 日间 测定浓度 (ng/ml) 精密度(CV%) 准确性(RE%) 测定浓度 (ng/ml) 精密度(CV%) 准确性(RE%) 尿嘧啶 10 10.2±0.38 3.74 2.58 10.12±0.78 7.70 1.18 20 20.63±1.21 5.87 3.15 19.97±1.35 6.74 −0.16 500 529.73±4.64 0.88 5.95 484.32±35.72 7.37 −3.12 1000 1093.33±25.10 2.30 9.33 1098.25±25.16 2.29 9.82 二氢尿嘧啶 10 10.32±0.71 6.86 3.18 10.28±0.65 6.37 2.77 20 19.98±2.19 10.95 −0.12 19.86±1.85 9.31 −0.72 500 517.51±10.69 2.07 3.50 515.66±10.36 2.01 3.13 1000 1079.83±17.91 1.66 7.98 1080.11±24.50 2.27 8.01  下载: 导出CSV
下载: 导出CSV
表 2 尿嘧啶和二氢尿嘧啶的基质效应和提取回收率
分析物 标识浓度(ng/ml) 基质效应 提取回收率 平均基质效应 CV(%) 平均回收率 CV(%) 尿嘧啶 1000 101.00 6.15 94.98 9.01 20 99.99 3.63 100.01 7.64 二氢尿嘧啶 1000 85.72 2.07 106.47 1.58 20 93.58 4.53 99.54 9.77
下载: 导出CSV
表 3 样品的稳定性(RE%)
分析物 冻融3次 室温放置6 h 置自动进样器24 h 低 高 低 高 低 高 尿嘧啶 100.71 98.34 93.49 106.60 108.20 107.99 二氢尿嘧啶 92.67 92.64 93.61 107.26 106.97 107.15
下载: 导出CSV
-
[1] SHARMA V, GUPTA S K, VERMA M. Dihydropyrimidine dehydrogenase in the metabolism of the anticancer drugs[J]. Cancer Chemother Pharmacol,2019,84(6):1157-1166. doi: 10.1007/s00280-019-03936-w [2] KOBUCHI S, AKUTAGAWA M, ITO Y, et al. Association between the pharmacokinetics of capecitabine and the plasma dihydrouracil to uracil ratio in rat: a surrogate biomarker for dihydropyrimidine dehydrogenase activity[J]. Biopharm Drug Dispos,2019,40(1):44-48. doi: 10.1002/bdd.2168 [3] AMSTUTZ U, FROEHLICH T K, LARGIADÈR C R. Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5-fluorouracil toxicity[J]. Pharmacogenomics,2011,12(9):1321-1336. doi: 10.2217/pgs.11.72 [4] DEENEN M J, TOL J, BURYLO A M, et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer[J]. Clin Cancer Res,2011,17(10):3455-3468. doi: 10.1158/1078-0432.CCR-10-2209 [5] SISTONEN J, BÜCHEL B, FROEHLICH T K, et al. Predicting 5-fluorouracil toxicity: DPD genotype and 5, 6-dihydrouracil: uracil ratio[J]. Pharmacogenomics,2014,15(13):1653-1666. doi: 10.2217/pgs.14.126 [6] ROBIN T, SAINT-MARCOUX F, TOINON D, et al. Automatic quantification of uracil and dihydrouracil in plasma[J]. J Chromatogr B Analyt Technol Biomed Life Sci,2020,1142:122038. doi: 10.1016/j.jchromb.2020.122038 [7] ANTUNES M V, RAYMUNDO S, CEZIMBRA DA SILVA A C, et al. Determination of endogenous concentrations of uracil and dihydrouracil in dried saliva spots by LC-MS/MS: method development, validation, and clinical application[J]. Ther Drug Monit,2019,41(3):383-390. doi: 10.1097/FTD.0000000000000615 [8] CHAVANI O, JENSEN B P, STROTHER R M, et al. Development, validation and application of a novel liquid chromatography tandem mass spectrometry assay measuring uracil, 5, 6-dihydrouracil, 5-fluorouracil, 5, 6-dihydro-5-fluorouracil, α-fluoro-β-ureidopropionic acid and α-fluoro-β-alanine in human plasma[J]. J Pharm Biomed Anal,2017,142:125-135. doi: 10.1016/j.jpba.2017.04.055 [9] JACOBS B A, ROSING H, DE VRIES N, et al. Development and validation of a rapid and sensitive UPLC-MS/MS method for determination of uracil and dihydrouracil in human plasma[J]. J Pharm Biomed Anal,2016,126:75-82. doi: 10.1016/j.jpba.2016.04.039 [10] ZHENG N Y, ZENG J N, JI Q C, et al. Bioanalysis of dried saliva spot (DSS) samples using detergent-assisted sample extraction with UHPLC-MS/MS detection[J]. Anal Chim Acta,2016,934:170-179. doi: 10.1016/j.aca.2016.05.057 [11] 肖力, 任斌, 陈小陆, 等. 高效液相色谱法测定人血浆中内源性尿嘧啶和二氢尿嘧啶含量[J]. 中国医院药学杂志, 2008, 28(2):112-114. doi: 10.3321/j.issn:1001-5213.2008.02.010 [12] CASNEUF V, BORBATH I, VAN DEN EYNDE M, et al. Joint Belgian recommendation on screening for DPD-deficiency in patients treated with 5-FU, capecitabine (and tegafur)[J]. Acta Clin Belg,2021:1-7. [13] DOLAT M, MACAIRE P, GOIRAND F, et al. Association of 5-FU therapeutic drug monitoring to DPD phenotype assessment may reduce 5-FU under-exposure[J]. Pharmaceuticals (Basel),2020,13(11):E416. doi: 10.3390/ph13110416 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5312
- HTML全文浏览量: 2382
- PDF下载量: 62
- 被引次数: 0
