

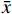
 下载:
下载:
 下载:
下载:

-
肾指海绵属(Reniochalina)海绵为寻常海绵纲(Demospongiae)软海绵目(Halichondrida)小轴海绵科(Axinellidae)的海洋多细胞动物。目前,针对该属海绵进行的化学成分研究主要包括甾类[1]、低极性化合物(如脂肪酸类、邻苯二甲酸类、烃类)[2]、炔醇[3]和环肽[4]等类型。其中,环肽reniochalistatins A-E由我们课题组获得,而环八肽reniochalistatin E因对人骨髓瘤细胞RPMI-8226的IC50值为4.90 μmol/L,已有两个团队采用不同策略对其完成全合成[5-6]。
为进一步高效挖掘肾指海绵中的潜在新型活性环肽,课题组基于环肽类化合物质谱响应度好,灵敏度高,二级质谱中存在明显的氨基酸单元碎裂的b和y特征离子,以质谱为导向,继续追踪分离该属海绵中的环肽类成分。最终,分离鉴定了3个环七肽(图1),并对它们进行了初步细胞毒活性评价。
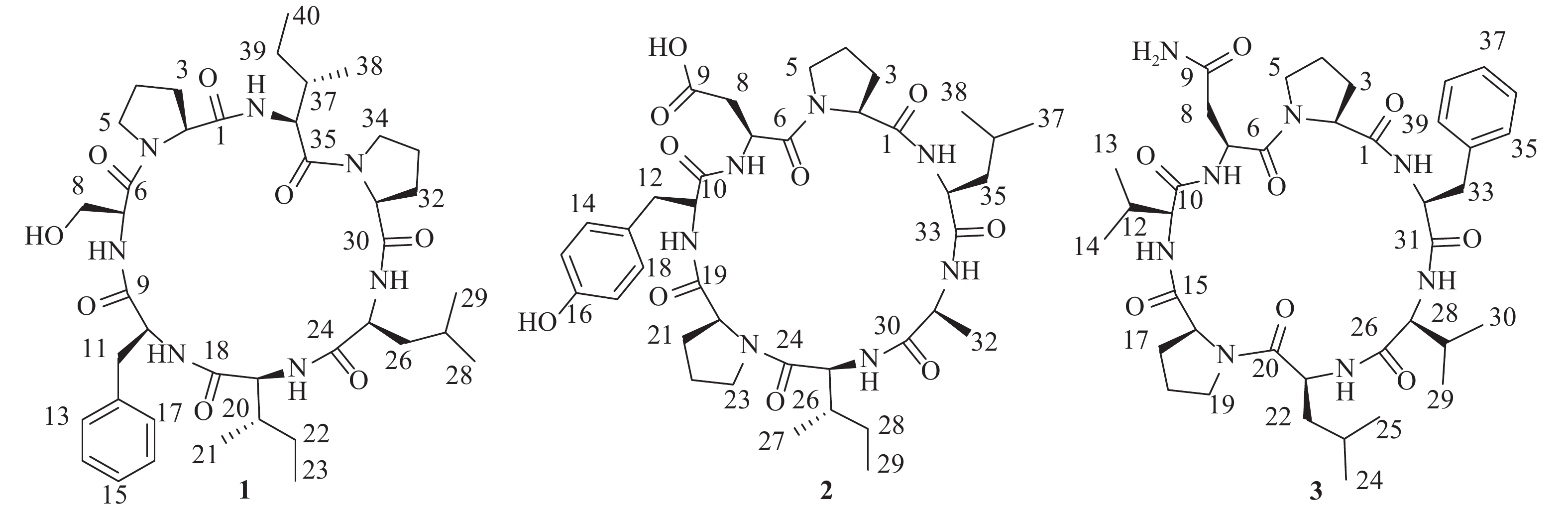
图 1 化合物1 ~ 3的化学结构
-
600、700 MHz核磁共振波谱仪(德国Bruker公司);Xevo G2-XS QTOF质谱仪(美国Waters公司);质谱引导的自动纯化系统(美国Waters公司);Acquity UPLC高效液相色谱仪(美国Waters公司);Interchim PuriFlash 450中压色谱仪(法国Interchim公司);SK5200H型超声仪(上海科导超声仪器有限公司);BT224S电子天平(德国Sartorius公司);EYELAN-1000型旋转蒸发仪(日本东京理化公司);低温高速离心机(美国Thermo Fisher公司);XBridge C18半制备型液相色谱柱(10 mm × 250 mm, 5 μm,美国Waters公司);ODS柱色谱填料(日本YMC公司);Sephadex-LH20柱色谱填料(瑞典Pharmacia公司);色谱级试剂(德国Merck公司);分析纯试剂(上海化学试剂公司);氘代试剂(美国Cambridge Isotope公司)。
-
海绵样本由课题组颜益珍老师于2021年4月采集自中国南海永乐群岛附近海域(25~30 m深度)。样本呈橙黄色,质地较硬。经课题组杨琪博士鉴定为Reniochalina属海绵。凭证标本(编号:212604)现保存于上海交通大学医学院附属仁济医院海洋药物实验室。
-
将干重78.5 g的海绵剪碎成块,先后加入等体积甲醇、二氯甲烷-甲醇(V/V = 1∶1)分别超声提取3~5次,每次30 min,合并提取液经减压浓缩得粗提物23.7 g。先将粗提物混悬于90%的甲醇水中,用等体积石油醚萃取3~5次,再将90%的甲醇水层加水稀释至60%,用等体积二氯甲烷萃取3~5次,合并萃取液经减压浓缩得二氯甲烷层浸膏1.2 g,其经LC-MS分析确认富含潜在的大分子量环肽。而60%的甲醇水层稀释至30%后再用乙酸乙酯萃取,经LC-MS检测已无大分子量环肽。故通过质谱导向的分步萃取,潜在的大分子量环肽主要分布于二氯甲烷层。
-
基于环肽类化合物分子量大的特点,通过两次Sephadex LH-20凝胶柱色谱分离,并采用质谱定位追踪的方式将潜在环肽富集。第一次以二氯甲烷-甲醇(V/V = 1∶1)为洗脱剂,二氯甲烷层浸膏经凝胶柱色谱分离得到组分Fr.A ~ Fr.C。第二次以正己烷-二氯甲烷-甲醇(V/V/V = 4∶5∶1)为洗脱剂,富含潜在大分子量环肽的Fr.B经凝胶柱色谱分离得到亚组分Fr.B1 ~ Fr.B5。其中,Fr.B2 ~ Fr.B4先后经中压ODS柱色谱分离(紫外监测波长设为210 nm,以甲醇-水为流动相,以流速15 ml/min在10 h内梯度洗脱10% ~ 100%),通过质谱定位追踪分别获得一系列精细组分。
上述精细组分经质谱引导的自动纯化系统制备获得环肽单体。质谱条件:质量扫描范围m/z 100~1250,锥孔电压 15 V,ES+模式下扫描采集数据。色谱条件:以半制备型C18柱为固定相,以乙腈-水(含0.1% 甲酸)为流动相,以流速6 ml/min进行梯度洗脱。其中,Fr.B2-27在32 min内线性梯度洗脱40%~48%获得化合物1(2.7 mg, tR=21 min);Fr.B4-5在52 min内线性梯度洗脱20%~23%获得化合物2(3.3 mg, tR=36 min);Fr.B2-11在32 min内线性梯度洗脱30%~40%获得化合物3(3.4 mg, tR=18 min)。
-
化合物1:无色结晶固体,茚三酮显色阴性。HRESIMS给出准分子离子峰m/z 768.4661 [M+H]+ (calcd 768.4660),确定其分子量为767。结合1H和13C-NMR谱确定其分子式为C40H61N7O8,计算不饱和度为14。1D NMR谱图给出典型的肽类化合物特征信号。其中,1H NMR谱显示有5个低场区的酰胺氨基质子信号(δH 9.45, 8.90, 8.71, 7.23, 7.16),7个α-次甲基质子信号(δH 4.69, 4.32, 4.16, 4.12, 4.03, 3.89, 3.51)。13C NMR谱提示有7个酰胺羰基碳信号(δC 171.6, 171.1, 170.9, 170.8, 170.6, 169.6, 168.5),7个α-次甲基碳信号(δC 60.7, 60.6, 59.2, 55.4, 55.1, 55.0, 54.1)。由此推断该化合物可能为七肽。结合2D NMR谱图,确定该七肽由两个脯氨酸残基、一个亮氨酸残基、两个异亮氨酸残基、一个苯丙氨酸残基和一个丝氨酸残基组成。残基连接顺序由HMBC、ROESY相关信号和ESI-MS/MS质谱数据确定,绝对构型由高级Marfey法确定[7-8]。综上,该化合物结构为cyclo-(L-Pro-L-Ser-L-Phe-L-Ile-L-Leu-L-Pro-L-Ile)。将核磁共振数据归属如下:1H NMR (700 MHz, DMSO-d6) δH 9.45 (1H, d, J = 8.4 Hz, 36-NH), 8.90 (1H, br s, 7-NH), 8.71 (1H, d, J = 6.6 Hz, 25-NH), 7.23 (1H, d, J = 10.3 Hz, 19-NH), 7.16 (1H, d, J = 4.8 Hz, 10-NH), 7.31 (2H, d, J = 7.5 Hz, H-13, H-17), 7.25 (2H, t, J = 7.5 Hz, H-14, H-16), 7.20 (1H, t, J = 7.3 Hz, H-15), 4.69 (1H, d, J = 7.8 Hz, H-2), 4.39 (1H, m, H-34a), 4.32 (1H, dd, J = 11.0, 8.4 Hz, H-36), 4.16 (1H, m, H-10), 4.12 (1H, t, J = 10.5 Hz, H-19), 4.03 (1H, t, J = 7.8 Hz, H-31), 3.89 (1H, t, J = 7.1 Hz, H-7), 3.57 (1H, m, H-34b), 3.51 (1H, m, H-25), 3.50 (1H, m, H-5a), 3.49 (1H, m, H-8a), 3.47 (1H, m, H-8b), 3.36 (1H, m, H-5b), 3.13 (1H, dd, J = 13.1, 2.8 Hz, H-11a), 2.84 (1H, dd, J = 13.0, 9.5 Hz, H-11b), 2.38 (1H, m, H-3a), 2.18 (1H, td, J = 12.7, 4.3 Hz, H-26a), 2.08 (1H, m, H-32a), 2.05 (1H, m, H-37), 2.01 (1H, m, H-33a), 1.92 (1H, m, H-3b), 1.90 (1H, m, H-4a), 1.79 (1H, m, H-33b), 1.73 (1H, m, H-32b), 1.55 (1H, m, H-26b), 1.53 (1H, m, H-39a), 1.52 (1H, m, H-4b), 1.43 (1H, m, H-20), 1.42 (2H, m, H-22a, H-27), 1.15 (1H, m, H-39b), 1.09 (1H, m, H-22b), 0.87 (3H, m, H-21), 0.85 (3H, m, H-29), 0.83 (6H, m, H-23, H-38), 0.82 (3H, m, H-28), 0.75 (3H, t, J = 7.3 Hz, H-40);13C NMR (175 MHz, DMSO-d6) δC 171.6 (C-30), 171.1 (C-35), 170.9 (C-9), 170.8 (C-1), 170.6 (C-24), 169.6 (C-18), 168.5 (C-6), 136.9 (C-12), 129.9 (C-13, C-17), 127.9 (C-14, C-16), 126.4 (C-15), 60.7 (C-31), 60.6 (C-2, C-8), 59.2 (C-19), 55.4 (C-7), 55.1 (C-10), 55.0 (C-36), 54.1 (C-25), 48.2 (C-34), 46.3 (C-5), 37.0 (C-11, C-26), 36.7 (C-20), 34.8 (C-37), 30.8 (C-3), 29.4 (C-32), 24.7 (C-27, C-33), 24.2 (C-22), 23.9 (C-39), 23.2 (C-29), 22.2 (C-4), 20.8 (C-28), 14.9 (C-40), 14.5 (C-23), 10.1 (C-38), 9.5 (C-21)。以上数据与文献[9]基本一致,故将化合物1鉴定为stylopeptide 1。
化合物2:无色无定形粉末,茚三酮显色阴性。HRESIMS给出准分子离子峰m/z 770.4102 [M+H]+ (calcd 770.4089),确定其分子量为769。结合1H和13C-NMR谱确定其分子式为C38H55N7O10,计算不饱和度为15。1D NMR谱图给出典型的肽类化合物特征信号。其中,1H NMR谱显示有5个低场区的酰胺氨基质子信号(δH 8.77, 8.43, 8.33, 8.03, 7.55),7个α-次甲基质子信号(δH 4.53, 4.38, 4.34, 4.28, 4.05, 4.03, 3.99)。13C NMR谱提示有7个酰胺羰基碳信号(δC 172.6, 172.4, 170.6, 170.5, 170.3, 170.0, 169.9),7个α-次甲基碳信号(δC 62.5, 60.4, 57.5, 56.9, 51.6, 49.9, 47.4)。据此推断该化合物可能为七肽。结合2D NMR谱图,确定该七肽由两个脯氨酸残基、一个亮氨酸残基、一个酪氨酸残基、一个天冬氨酸残基、一个丙氨酸残基和一个异亮氨酸残基组成。残基连接顺序由HMBC、ROESY相关信号和ESI-MS/MS质谱数据确定,绝对构型由高级Marfey法确定。综上,该化合物结构为cyclo-(L-Pro-L-Asp-L-Tyr-L-Pro-L-Ile-L-Ala-L-Leu)。将核磁共振数据归属如下:1H NMR (700 MHz, DMSO-d6) δH 8.77 (1H, br s, 31-NH), 8.43 (1H, d, J = 7.5 Hz, 11-NH), 8.33 (1H, br s, 25-NH), 8.03 (1H, s, 7-NH), 7.55 (1H, d, J = 6.5 Hz, 34-NH), 6.97 (2H, d, J = 8.1 Hz, H-14, H-18), 6.66 (2H, d, J = 8.1 Hz, H-15, H-17), 4.53 (1H, m, H-7), 4.38 (1H, m, H-31), 4.34 (1H, m, H-20), 4.28 (1H, d, J = 7.5 Hz, H-34), 4.05 (1H, m, H-11), 4.03 (1H, m, H-2), 3.99 (1H, m, H-25), 3.63 (1H, m, H-5a), 3.56 (1H, m, H-5b), 3.26 (2H, m, H-8a, H-23a), 2.99 (1H, m, H-12a), 2.88 (1H, m, H-12b), 2.83 (1H, m, H-8b), 2.59 (1H, m, H-23b), 2.22 (1H, m, H-3a), 2.10 (1H, m, H-21a), 1.87 (1H, m, H-4a), 1.77 (1H, m, H-4b), 1.68 (1H, m, H-35a), 1.67 (2H, m, H-21b, H-26), 1.58 (1H, m, H-35b), 1.57 (1H, m, H-3b), 1.55 (1H, m, H-36), 1.54 (1H, m, H-22a), 1.53 (1H, m, H-28a), 1.24 (1H, m, H-22b), 1.16 (3H, d, J = 6.6 Hz, H-32), 1.10 (1H, m, H-28b), 0.86 (3H, d, J = 5.8 Hz, H-37), 0.82 (3H, m, H-38), 0.80 (6H, m, H-27, H-29); 13C NMR (175 MHz, DMSO-d6) δC 172.6 (C-1), 172.4 (C-24), 172.1 (C-9), 170.6 (C-19), 170.5 (C-10), 170.3 (C-6), 170.0 (C-33), 169.9 (C-30), 155.9 (C-16), 129.6 (C-14, C-18), 127.8 (C-13), 115.1 (C-15, C-17), 62.5 (C-2), 60.4 (C-20), 57.5 (C-31), 56.9 (C-34), 51.6 (C-7), 49.9 (C-11), 47.4 (C-25), 47.0 (C-5), 45.7 (C-23), 40.4 (C-35), 36.1 (C-8), 35.4 (C-12, C-26), 30.4 (C-3), 29.5 (C-21), 25.1 (C-4), 24.7 (C-36), 24.3 (C-28), 22.9 (C-37), 21.4 (C-38), 20.9 (C-22), 15.1 (C-32), 14.9 (C-29), 10.9 (C-27)。以上数据与文献[10]基本一致,故将化合物2鉴定为hymenamide D。
化合物3:无色无定形粉末,茚三酮显色阴性。HRESIMS给出准分子离子峰:m/z 767.4468 [M+H]+ (calcd 767.4456),确定其分子量为766。结合1H和13C-NMR谱确定其分子式为C39H58N8O8,计算不饱和度为15。1D NMR谱图给出典型的肽类化合物特征信号。其中,1H NMR谱显示有5个低场区的酰胺氨基质子信号(δH 8.80, 8.13, 7.98, 7.95, 7.25),7个α-次甲基质子信号(δH 4.65, 4.28, 4.24, 4.19, 4.14, 3.89, 3.83)。13C NMR谱提示有7个酰胺羰基碳信号(δC 171.4, 171.1, 170.9, 170.3, 170.2, 170.1, 169.6),7个α-次甲基碳信号(δC 62.3, 61.5, 60.2, 57.1, 55.2, 51.0, 49.3)。据此推断该化合物可能为七肽。结合ESI-MS/MS质谱数据确定其氨基酸残基组成为两个脯氨酸残基、一个亮氨酸残基、一个苯丙氨酸残基、一个天冬酰胺残基和两个缬氨酸残基,残基连接顺序为cyclo-(Pro-Asn-Val-Pro-Leu-Val-Phe)。通过高级Marfey法确定所有氨基酸残基均为L构型。将核磁共振数据归属如下:1H NMR (600 MHz, DMSO-d6) δH 8.80 (1H, d, J = 5.4 Hz, 21-NH), 8.13 (1H, d, J = 9.5 Hz, 32-NH), 8.01 (1H, s, 9-NH2), 7.98 (1H, d, J = 8.5 Hz, 11-NH), 7.95 (1H, d, J = 6.1 Hz, 7-NH), 7.28 (1H, m, 9-NH2), 7.25 (3H, m, 27-NH, H-35, H-39), 7.18 (1H, d, J = 7.4 Hz, H-37), 7.14 (2H, d, J = 7.6 Hz, H-36, H-38 ), 4.65 (1H, m, H-7), 4.28 (1H, d, J = 7.7 Hz, H-32), 4.24 (1H, m, H-16), 4.19 (1H, m, H-21), 4.14 (1H, t, J = 8.6 Hz, H-27), 3.89 (1H, t, J = 8.6 Hz, H-2), 3.83 (1H, t, J = 7.9 Hz, H-11), 3.75 (1H, m, H-5a), 3.46 (2H, m, H-5b, H19a), 3.26 (1H, m, H19b), 3.06 (1H, m, H-8a), 2.94 (3H, m, H-8b, H33a, H33b), 2.34 (1H, m, H-17a), 2.19 (2H, m, H-3a, H12), 2.02 (1H, m, H-28), 1.96 (1H, m, H-18a), 1.85 (1H, m, H-4a), 1.71 (2H, m, H-18b, H-23), 1.61 (1H, m, H-22a), 1.46 (1H, m, H-4b), 1.23 (1H, m, H-17b), 1.19 (1H, m, H-22b), 0.99 (1H, m, H-3b), 0.92 (3H, d, J = 6.7 Hz, H-13), 0.89 (3H, d, J = 6.8 Hz, H-14), 0.86 (6H, d, J = 6.7 Hz, H-29, H-30), 0.84 (3H, d, J = 6.7 Hz, H-25), 0.79 (3H, d, J = 6.4 Hz, H-24); 13C NMR (150 MHz, DMSO-d6) δC 172.5 (C-9), 171.4 (C-26), 171.1 (C-20), 170.9 (C-31), 170.3 (C-1), 170.2 (C-15), 170.1 (C-10), 169.6 (C-6), 138.4 (C-34), 128.9 (C-35, C-39), 128.1 (C-36, C-38), 126.2 (C-37), 62.3 (C-2), 61.5 (C-11), 60.2 (C-16), 57.1 (C-27), 55.2 (C-32), 51.0 (C-21), 49.3 (C-7), 47.6 (C-5), 45.8 (C-19), 37.9 (C-22), 37.0 (C-33), 35.5 (C-8), 30.5 (C-17), 30.1 (C-28), 29.5 (C-12), 29.2 (C-3), 25.1 (C-4), 24.2 (C-23), 23.3 (C-24), 21.3 (C-18), 20.6 (C-25), 19.7 (C-13), 18.7 (C-14, C-29), 18.3 (C-30)。以上数据与文献[11]基本一致,故将化合物3鉴定为axinastatin 2。
-
采用CCK-8法初步评估化合物对6种人肿瘤细胞株(A2780、HCT-8、HepG2、NCI-H460、SW480、PC-9)的体外细胞毒活性。主要步骤为:取对数生长期的细胞制成细胞悬液,约4×103个细胞/孔接种至96孔板内,每组设3个复孔。过夜培养后,各株细胞分别加入20 μmol/L化合物,并设对照组(顺铂)和空白组(DMSO),继续孵育48 h。随后,避光情况下每孔加入10 µl CCK-8溶液。孵育0.5 ~ 2 h后,450 nm处测其吸光度,每株细胞测3次以上,并计算其细胞活力(%)。对于肿瘤细胞生长抑制率大于50%的化合物,进一步测试其活性剂量依赖关系。结果显示,化合物1对NCI-H460、HepG2、PC-9、HCT-8、A2780和SW480均具细胞毒性,IC50值分别为6.09、9.31、13.24、14.31、14.38和17.26 μmol/L。
-
环肽具有许多独特的生化和治疗特性,目前已有抗生素类达托霉素、止痛剂齐考诺肽、抗肿瘤药帕瑞肽等环肽类药物获批用于临床[12]。海绵是环肽类化合物的重要多产来源,然而,其体内环肽因含量低微、紫外吸收不佳、核磁灵敏度差等,特异识别和定向获取极富挑战。将质谱高灵敏度、高准确度和高选择性的技术优势与环肽独特的质谱碎裂模式有机结合,有助于提高环肽发现的通量和选择性。
本研究以质谱为导向,追踪分离肾指海绵中的潜在新颖活性环肽,分离鉴定出3个环七肽,它们均为首次从该属海绵分离获得。此外,本研究还发现化合物1对多种人肿瘤细胞株具有中等抑制活性。而有趣的是,早期由George R. Pettit课题组分离获得的源于Stylotella属和Phakellia属海绵的同一化合物1,其抑制P388小鼠白血病细胞生长的能力却相差十倍以上[9, 13]。这可能与天然来源环肽能与某些仅生物方法才能检测到的微量强活性抗肿瘤物结合[9, 13],或不同溶剂环境致使环肽分子构象发生改变有关[14]。化合物2先后发现于Hymeniacidon属[10]和Stylissa属[15-16]海绵,对多种肿瘤细胞株开展的活性试验显示其无明显细胞毒性。化合物3此前发现于Axinella属海绵,其对小鼠P388细胞和一系列人肿瘤细胞具有较强的体外细胞毒性[11]。
不同种属的海绵能够产生相同类型的环肽分子,同一类型的环肽分子表现出不同的细胞毒活性,可能和与海绵共生互作的微生物相关。本研究进一步丰富了肾指海绵中环肽类化合物的多样性,也为研究特征结构类型的海洋天然产物提供了新思路、新方法。
Mass spectrometry-guided study on cyclic peptides from sponge Reniochalina sp.
-
摘要:
目的 以质谱为导向对肾指海绵Reniochalina sp.中的环肽类成分进行研究。 方法 采用质谱引导的程序性分离手段定向追踪并分离纯化海绵中的环肽类成分;通过理化常数测定、波谱数据比对确定化合物结构;利用CCK-8法对化合物进行初步细胞毒活性评价。 结果 从肾指海绵Reniochalina sp.中分离获得3个环肽类化合物,分别鉴定为stylopeptide 1 ( 1 )、hymenamide D ( 2 )、axinastatin 2 ( 3 )。化合物 1 对6种人肿瘤细胞株具有细胞毒性,IC50值范围为6.09 ~ 17.26 μmol/L。 结论 化合物 1 ~ 3 首次分离自Reniochalina属海绵,化合物 1 是细胞毒性环七肽。 Abstract:Objective To study the cyclic peptides from sponge Reniochalina sp. under the guidance of mass spectrometry. Methods Mass spectrometry-guided procedural separation methods were used to track and isolate the cyclic peptides from the sponge genus Reniochalina. The structures of compounds were elucidated by the determination of physicochemical parameters and comparison of spectroscopic data. The preliminary cytotoxic activity of compounds was assessed by the Cell Counting Kit-8 (CCK-8) method. Results Three cyclic peptides were isolated from the sponge Reniochalina sp. and identified as stylopeptide 1 ( 1 ), hymenamide D ( 2 ) and axinastatin 2 ( 3 ). Compound 1 exhibited cytotoxicity against six human cancer cell lines with IC50 values ranging from 6.09 to 17.26 μmol/L. Conclusion Compound 1 - 3 were isolated from Reniochalina sp. for the first time, and compound 1 was a cytotoxic cyclic heptapeptide. -
Key words:
- Reniochalina sp. /
- chemical constituent /
- cyclic peptide /
- structure identification
-
在全球范围内,缺血性脑卒中(IS)仍然是导致死亡和严重残疾的最主要原因之一[1]。在IS的治疗过程中,除因出血风险而存在禁忌外,几乎所有IS的患者都应给予抗血小板药物进行二次卒中预防[2], 阿司匹林联合氯吡格雷的双重抗血小板治疗在IS发病早期发挥着重要作用[3]。然而临床应用中,脑血管病患者在服用氯吡格雷药物治疗期间存在个体差异,一些患者在服药后出现了病情反复发作或者病情加重的现象,称为氯吡格雷抵抗(CR)[4]。研究表明,导致氯吡格雷抵抗的因素是多种多样的,尚无统一的结论。包括性别[5-6]、年龄[6]、BMI[6-7]、糖尿病[5,8]、高脂血症[8]、高同型半胱氨酸[5,8]、药物间相互作用[8](钙离子拮抗剂、质子泵抑制剂)、遗传基因多态性[9-10](参与氧化代谢的相关基因CYP2C19、与其活化有关基因ABCB1)等均会导致该药物出现抵抗现象。但亦有部分研究报道得出相反结论[11]。且研究主要集中于心血管领域,尤其是经皮冠状动脉介入(PCI)术后患者,而针对IS患者发生CR影响因素的报道相对较少。氯吡格雷属于噻吩吡啶类化学衍生物,需要在多种酶的参与下才能够生成有活性的化合物,该活性代谢产物与血小板上的二磷酸腺苷(ADP)受体P2Y12共价特异性地结合,减少ADP所刺激的血小板凝聚,阻断ADP诱导的糖蛋白与纤维蛋白原PIIb/IIIa 受体的结合,发挥抗栓的效应[12-13]。故本研究利用血栓弹力图(TEG)测定ADP数值,对脑卒中患者的基线资料及CYP2C19基因多态性进行统计分析,评估产生CR现象的影响因素,为IS临床个体化用药、预测卒中复发风险提供精准医疗方案。
1. 材料和方法
1.1 研究对象
本研究利用HIS系统选取2021年4月至2022年7月在中部战区总医院神经内科住院治疗的IS患者。(1)纳入标准:①符合《中国脑急性缺血性脑卒中诊治指南》[14]或IS的相关诊断,且经CT或MRI影像学等证实;②年龄均>18岁;③未进行静脉溶栓或取栓治疗;④均采用氯吡格雷联合阿司匹林进行抗血小板治疗。(2)排除标准:①存在出血性疾病或颅内感染者;②合并恶性肿瘤或严重心、肝、肺、肾脏器官功能衰竭者;③有认知障碍者;④依从性差者;⑤对本研究药物过敏者。该研究已获得中部战区总医院医学伦理委员会审查批准,审查号:[2021]025-01。
1.2 治疗方法
所有入组患者入院后均给予改善循环(银杏达莫)、稳定血斑块(阿托伐他汀)、护胃(泮托拉唑)等常规治疗。在常规治疗基础上给予阿司匹林(拜耳医药保健有限公司,国药准字:J20171021,规格:100 mg/片)100 mg每日一次和硫酸氢氯吡格雷[赛诺菲(杭州)制药有限公司,国药准字:H20056410,规格:75 mg/片]75 mg 每日一次,均为3个月一个疗程。同时根据基础疾病给予降血压、降血糖及调血脂治疗。
1.3 资料收集
收集所有入组患者的基线资料,包括姓名、性别、年龄、既往有无高血压病、糖尿病、冠心病、高脂血症、吸烟史(戒烟小于5年)、饮酒史(戒酒小于5年)、既往有无缺血性脑卒中病史及患者住院期间合并用药等。记录患者在住院后次日清晨空腹采集的静脉血各项检验指标,包括血常规、肝肾功能、凝血功能等。
1.4 TEG及相关参数测定
患者在服用阿司匹林联合氯吡格雷药物7~14 d,空腹采集静脉血,分别注射入柠檬酸钠抗凝管1和肝素抗凝管2中。利用TEG-5000型TEG凝血分析仪(美国Haemoscope公司)检测柠檬酸-高岭土样品,得出ADP抑制率。将血小板聚集抑制率>30%定义为氯吡格雷非抵抗,血小板聚集抑制率≤30%定义为氯吡格雷抵抗[15]。
1.5 CYP2C19 基因检测
患者入院后次日清晨空腹抽取静脉血4 ml于EDTA抗凝管中,利用全自动杂交仪系统(BR-526-24,上海百傲科技公司)对CYP2C19*1、CYP2C19*2及CYP2C19*3位点进行检测。该检测过程主要包括全血DNA的提取、PCR扩增、芯片杂交显色,利用基因芯片识读仪自动读取结果。根据CYP2C19基因多态性进行药物代谢分组[16],即:快代谢组:EM,CYP2C19*1*1,中代谢组:IM,CYP2C19*1*2、CYP2C19*1*3,慢代谢组:PM,CYP2C19*2*2、CYP2C19*2*3、CYP2C19*3*3。
1.6 统计学方法
采用SPSS 26.0统计软件进行数据分析,计量资料服从正态分布,采用(
$\bar x $ ±s)表示,组间比较采用独立样本t检验。若计量资料属于非正态分布,则以中位数和四分位数[M(P25,P75)]表示,组间比较采用Mann-Whitney U检验。计数资料以例数和百分比[例(%)]表示,组间比较采用χ2检验,将IS患者发生氯吡格雷抵抗的影响因素进一步进行多因素Logisti回归分析,以P<0.05为差异有统计学意义。2. 结果
2.1 基线资料分析
本研究共纳入缺血性脑卒中患者202例,年龄35~86岁,其中,CR组87例,NCR组115例,CR组患者合并糖尿病的比例明显高于NCR组,差异有统计学意义(P<0.05)。在性别、年龄、吸烟、饮酒、既往有无缺血性脑卒中、高血压病、冠心病、高脂血症及合并用药方面,两组间差异均无统计学意义(P>0.05),见表1。
表 1 抵抗组和非抵抗组患者基线资料比较基本信息参数 CR组(n=87,%) NCR组(n=112,%) x2/t P 年龄 64.52±1.07 62.39±0.97 −1.47 0.14 性别(男) 65(74.7) 86(74.8) 0.00 0.99 吸烟史 43(49.4) 56(48.7) 0.11 0.92 饮酒史 37(42.5) 41(35.7) 0.99 0.32 IS既往史 16(18.4) 22(19.1) 0.02 0.89 高血压 83(95.4) 106(92.2) 0.41 0.53 糖尿病 35(40.2) 22(19.6) 10.15 0.03 冠心病 7(8.0) 10(8.7) 0.03 0.87 高脂血症 40(46.0) 56(48.7) 0.15 0.70 合并用药 质子泵抑制剂 61(70.1) 84(73.0) 0.21 0.67 丁苯酞注射液 67(77.0) 83(72.2) 0.61 0.44 2.2 实验室检验指标分析
氯吡格雷抵抗组血常规的白细胞计数水平高于非抵抗组,且组间差异有统计学意义(P<0.05)。两组患者红细胞、血红蛋白、血小板计数、凝血酶原时间、D-二聚体、血糖、总胆固醇、甘油三酯、高密度脂蛋白胆固醇、低密度脂蛋白、超敏C反应蛋白、血同型半胱氨酸、直接胆红素、总胆红素、谷草转氨酶、谷丙转氨酶的组间差异均无统计学意义(P>0.05),见表2。
表 2 两组患者实验室检测指标比较检测项目 抵抗组(n=87) 非抵抗组(n=115) 检验值 P 白细胞 6.70(5.60,8.40) 6.40(5.30,7.30) 2.14b 0.03 红细胞 4.56±0.34 4.44±0.05 −0.40a 0.69 血红蛋白 131(119,139) 134(120,143) −1.15b 0.11 血小板 187(148,227) 206(138,232) −1.62b 0.08 凝血酶原时间 11.21±0.65 11.18±0.85 −0.32a 0.92 D-二聚体 0.30±0.07 0.29±0.09 −0.78a 0.44 血糖 6.03±0.22 6.45±0.21 1.33a 0.18 总胆固醇 4.37(3.72,5.04) 4.32(3.73,5.05) −0.19b 0.85 甘油三酯 1.81±0.13 1.75±0.11 −0.36a 0.72 高密度脂蛋白 1.19(0.99,1.35) 1.14(1.00,1.32) −1.07b 0.29 低密度脂蛋白 2.41(1.94,2.80) 2.27(1.87,2.75) −0.50b 0.62 超敏C反应蛋白 1.91(1.04,5.40) 1.76(0.84,3.50) 1.65b 0.09 血同型半胱氨酸 17.32±1.18 15.81±0.61 −1.22a 0.23 直接胆红素 3.32±1.25 3.79±0.33 1.47a 0.14 总胆红素 13.33±0.47 13.71±0.42 0.60a 0.54 谷草转氨酶 23.00(19.00,28.00) 22.00(18.00,27.00) −0.89b 0.37 谷丙转氨酶 22.00(17.00,31.00) 21.00(15.00,28.79) −1.42b 0.15 注: a为 χ2 值,b为z值。 2.3 CYP2C19基因多态性分布
入选的202名患者中,CYP2C19 *1、CYP2C19 *2及CYP2C19*3等位基因的频率分别为62.26%、33.17%及4.21%,所有基因位点均符合Hardy-Weinberg平衡定律,差异无统计学意义(P>0.05),表明该群体具有代表性,详见表3。
表 3 202例IS患者 CYP2C19 基因型及等位基因的频率分布CYP2C19等位基因 等位基因数/个 基因频率/% CYP2C19 *1 253 62.62 CYP2C19 *2 134 33.17 CYP2C19 *3 17 4.21 2.4 不同CYP2C19代谢类型患者发生氯吡格雷抵抗的比较
CYP2C19 IM组患者发生氯吡格雷抵抗的比例显著高于EM组和PM组,差异具有统计学意义(P>0.05),见表4。
表 4 不同CYP2C19代谢类型的CR发生率比较(n,%)CYP2C19
代谢类型氯吡格雷
抵抗[n(%)]非氯吡格雷
抵抗[n(%)]χ2 p EM 24(27.59) 56(48.70) 9.95 0.007 IM 50(57.47) 43(37.39) PM 13(14.94) 16(13.91) 2.5 CYP2C19不同基因型血小板抑制率的比较
3组IS患者中ADP抑制率均不符合正态分布,因此采用非参数检验。EM组ADP抑制率的中位数为39.6%,IM组为26.10%,PM组为31.95%。3组血小板抑制率中位数差异具有统计学意义(P<0.05);两两比较,IM组的血小板抑制率明显低于EM组,差异具有统计学意义(P<0.05);其他组间差异无统计学意义(P>0.05),详见表5。
表 5 不同基因型血小板抑制率的比较CYP2C19基因分组 基因型数/个 血小板抑制率/% EM 39.60(26.20,53.95) CYP2C19*1*1 80 IM 26.10(15.20,53.60)* CYP2C19*1*2 81 CYP2C19*1*3 12 PM 31.95(20.65,65.40) CYP2C19*2*2 24 CYP2C19*2*3 5 χ2 6.31 P 0.04 注:3组间比较采用 Kruskal-Wallis H检验;两两比较采用 Mann- Whitney U检验;* P<0.05,与EM组比较。 2.6 发生氯吡格雷抵抗的危险因素分析
以发生CR作为因变量,将上述研究中存在统计学差异(P<0.05)的项目作为自变量进行赋值,包括合并糖尿病(未合并糖尿病者赋值0,合并糖尿病者赋值1)、CYP2C19代谢类型(IM赋值1,其余赋值0),白细胞计数为连续型数值变量,纳入二元Logistic回归模型中进行分析。结果显示,合并糖尿病、高白细胞计数水平、CYP2C19中代谢型为发生氯吡格雷抵抗的独立危险因素,见表6。
表 6 发生氯吡格雷抵抗危险因素的Logistic回归分析结果因素 B SE Wald P OR 95%CI 合并糖尿病 0.595 0.227 4.496 0.026 0.851 (0.707,1.191) 白细胞计数 0.211 0.076 7.774 0.005 1.235 (1.065,1.432) CYP2C19
中代谢型0.901 0.291 9.565 0.002 0.368 (0.197,0.686) 3. 讨论
氯吡格雷是继阿司匹林后在心脑血管中被广泛使用的抗血小板药物。有报道表明氯吡格雷联合阿司匹林在治疗缺血性脑卒中疗效优于单独使用阿司匹林,且不会增加脑卒中患者发生出血的风险[3,17]。在临床应用中仍有一些患者在服用氯吡格雷后,不能够抑制ADP诱导的血小板凝聚,也称为氯吡格雷抵抗或者氯吡格雷低反应性[4]。目前国际上关于氯吡格雷抵抗的诊断并无确切的标准[18],有文献称化学比浊法属于比较经典的血小板功能检测方法,但是由于在操作过程中步骤比较繁多且尚无统一的标准化流程,现已逐渐被其它检测方法所取代[19]。有国外研究报道将床旁快速血小板分析(Verify Now)检测系统的检测结果定义为“金标准”,但是由于该检测方法成本较高,因此在国内临床中并没有被大规模应用。而现在国内较常使用的抗血小板效果的监测方法有TEG、血小板分析仪、PL-11等,TEG检测血小板功能操作便捷、迅速、受外界影响较小等优点,现已在临床中被广泛使用[20]。
氯吡格雷本身属于无活性的前体药物,需在多种酶的参与下生成有活性的化合物,才能发挥抑制血小板聚集作用。细胞色素P450酶参与了两步氧化代谢过程。国外有研究报道[21]CYP2C19*2、CYP2C19*3等位基因的突变主要以亚洲人为主,发生在非洲和欧洲的突变率较低,携带CYP2C19*2、CYP2C19*3基因的突变可降低细胞色素P450酶的活性,使得活性代谢物的水平减少,从而导致该药物的临床疗效不佳,所以IM和PM组氯吡格雷抗血小板活性会受到CYP2C19基因多态性的影响。本研究结果显示,CYP2C19 IM组患者发生氯吡格雷抵抗的比例显著高于EM组和PM组,血小板抑制率明显低于EM组,差异均具有统计学意义,而PM组血小板抑制率虽然低于EM组,但差异无统计学意义,可能是PM组(14.36%)人群占比显著低于IM组(46.04%)和EM组(39.60%)造成的。
目前已有研究表明糖尿病患者在服用氯吡格雷后血小板抑制率较差[22-23],但其机制尚不明确。在小鼠模型中的研究结果表明,高血糖可以通过下调CYP酶影响氯吡格雷药物活化,从而导致抗血小板浓度降低[24]。Angiolillo等[25]针对冠心病合并2型糖尿病患者诱导的血小板抑制率减弱的研究中,解释其原因可能是长期的高血糖水平使血小板信号通路上调降低了抗血小板药物的药效,从而导致药物抵抗的发生。此外,糖尿病会影响肝脏代谢与胃肠道吸收,这可能会影响药物的吸收和代谢[26]。本研究结果也显示,合并糖尿病是发生氯吡格雷抵抗的独立风险因素。
本研究结果还表明高白细胞计数可能是IS患者发生CR的独立危险因素(OR=1.235,95% CI:1.065,1.432),但目前在IS患者中有关白细胞计数与CR之间关系的研究较少。有研究发现炎症标志物升高增加心血管事件的风险,血小板聚集和炎性因子之间的相互作用在急性冠脉综合征中起着非常重要的作用,而炎症调节越来越被认为是当代的治疗靶点,接受氯吡格雷联合阿司匹林双重抗血小板治疗的急性冠状动脉综合征的患者中,白细胞计数与临床疗效密切相关[27]。也有研究者在冠心病患者中发现白细胞中的3个长链非编码RNA与CR的发生有关[28]。IS随着细胞死亡和脑组织受损,分子危险信号通过激活更多的小胶质细胞和浸润白细胞进行前馈反应,释放更多的具有促炎作用的细胞因子。炎症加剧了脑细胞受损程度,使外周血中释放更多的细胞因子,这些细胞因子参与了脑卒中的进展,影响其预后[29-30]。因此,在临床中,高白细胞计数患者服用氯吡格雷时应警惕CR的发生。
本研究的局限性主要包括以下几点:①样本量相对较少;②TEG检测过程中受血凝速度、红细胞聚集状态等因素影响;③患者服药7~14 d只检测1次ADP抑制率,不能反应整体水平;④未直接检测血浆氯吡格雷及其活性代谢物水平。上述因素可能对研究结果产生偏倚,今后还需要进一步的验证。
-
[1] 薛松. 海绵活性天然产物分离及结构确定[D]. 大连: 中国科学院研究生院(大连化学物理研究所), 2003. [2] 闫小燕, 靳艳, 虞星炬, 等. 气相色谱-质谱分析新种海绵Reniochalina sp. 中的低极性组分[J]. 色谱, 2004, 22(6):652-654. doi: 10.3321/j.issn:1000-8713.2004.06.021 [3] LEE H S, LEE J H, WON H, et al. Identification of novel acetylenic alcohols and a new dihydrothiopyranone from the tropical sponge Reniochalina sp.[J]. Lipids,2009,44(1):71-75. doi: 10.1007/s11745-008-3249-3 [4] ZHAN K X, JIAO W H, YANG F, et al. Reniochalistatins A-E, cyclic peptides from the marine sponge Reniochalina stalagmitis[J]. J Nat Prod,2014,77(12):2678-2684. doi: 10.1021/np5006778 [5] ATINO A, BACA G, WEERAMANGE C, et al. Total synthesis of reniochalistatin E[J]. J Nat Prod,2017,80(12):3234-3240. doi: 10.1021/acs.jnatprod.7b00656 [6] ZHOU R, SUN Y G, LI H B, et al. Synthesis and biological evaluation of reniochalistatins A-E and a reniochalistatin E analogue[J]. ChemMedChem,2018,13(20):2202-2207. doi: 10.1002/cmdc.201800529 [7] FUJII K, IKAI Y, MAYUMI T, et al. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: elucidation of limitations of marfey’s method and of its separation mechanism[J]. Anal Chem,1997,69(16):3346-3352. doi: 10.1021/ac9701795 [8] FUJII K, IKAI Y, OKA H, et al. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: combination of marfey’s method with mass spectrometry and its practical application[J]. Anal Chem,1997,69(24):5146-5151. doi: 10.1021/ac970289b [9] PETTIT G R, SRIRANGAM J K, HERALD D L, et al. Isolation and crystal structure of stylopeptide 1, A new marine Porifera cycloheptapeptide[J]. J Org Chem,1995,60(25):8257-8261. doi: 10.1021/jo00130a027 [10] TSUDA M, SHIGEMORI H, MIKAMI Y, et al. Hymenamides C~E, new cyclic heptapeptides with two proline residues from the Okinawan marine sponge hymeniacidon sp.[J]. Tetrahedron,1993,49(31):6785-6796. doi: 10.1016/S0040-4020(01)80422-1 [11] PETTIT G R, GAO F, CERNY R L, et al. Antineoplastic agents. 278. Isolation and structure of axinastatins 2 and 3 from a western Caroline Island marine sponge[J]. J Med Chem,1994,37(8):1165-1168. doi: 10.1021/jm00034a014 [12] ZHANG H Y, CHEN S Y. Cyclic peptide drugs approved in the last two decades (2001-2021)[J]. RSC Chem Biol,2022,3(1):18-31. doi: 10.1039/D1CB00154J [13] PETTIT G R, TAYLOR S R. Synthesis of the marine sponge cycloheptapeptide stylopeptide 1[J]. J Org Chem,1996,61(7):2322-2325. doi: 10.1021/jo951986b [14] 刘晓庆. 生物大分子序列、结构及活性的计算模拟[D]. 大连: 大连理工大学, 2009. [15] AFIFI A H, EI-DESOKY A H, KATO H, et al. Carteritins A and B, cyclic heptapeptides from the marine sponge Stylissa carteri[J]. Tetrahedron Lett,2016,57(11):1285-1288. doi: 10.1016/j.tetlet.2016.02.031 [16] SUN J Y, CHENG W, DE VOOGD N J, et al. Stylissatins B-D, cycloheptapeptides from the marine sponge Stylissa massa[J]. Tetrahedron Lett,2016,57(38):4288-4292. doi: 10.1016/j.tetlet.2016.08.024 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3714
- HTML全文浏览量: 1105
- PDF下载量: 12
- 被引次数: 0
