

 下载:
下载:
 下载:
下载: 


-
近年来,我国药品审评审批制度逐步完善,2005年原国家食品药品监督管理局发布《药品特别审批程序》,对突发公共事件亟需的应急药品可进行特别审批[1]。2016年原国家食品药品监督管理总局发布的《总局关于解决药品注册申请积压实行优先审评审批的意见》[2],首次明确了优先审评审批的范围、程序和工作要求。2020年国家药品监督管理局发布《突破性治疗药物审评工作程序(试行)》等3个文件的公告[3],进一步明确了3种药品加快审批途径。这一系列应急审批程序及加快审评工作规范的发布,对于应急药品供应、创新药物研发具有重要意义:①药品审批效率不断提高,创新药的研发时间明显缩短,适用患者可尽早获得需要的药物治疗;②激励创新药研发机构加大投入并提高创新能力;③在面对诸如新型冠状病毒肺炎(COVID-19)疫情时,开辟绿色通道,相关疫苗及治疗药物迅速上市,在抗击疫情过程中发挥了重要作用。但总体来说,我国的药品应急审批制度起步较晚,实践过程中还在不断摸索,该研究通过分析对比我国与美国、欧盟、日本等国家与地区的药品应急审批制度,探讨进一步优化我国药品应急审批制度的思路和措施。
-
国家市场监督管理总局2020年1月发布的《药品注册管理办法》[4],将我国药品加快上市注册程序分为突破性治疗、附条件批准、优先审评审批和特别审批程序。该4种程序的适用范围和审批阶段见表1。
表 1 我国药品应急审批程序对比
名称 适用范围 申请阶段 突破性治疗药物 在药物临床试验期间,用于防治严重危及生命或严重影响生存质量的疾病且尚无有效防治手段或与现有治疗手段相比具有明显临床优势的创新药或改良新药等 在Ⅰ、Ⅱ期临床试验阶段,通常不晚于Ⅲ期临床试验开展前 附条件批准 治疗严重危及生命且尚无有效治疗手段的疾病的药品,药物临床试验已有数据证实疗效并能预测其临床价值的;公共卫生方面急需的药品,药物临床试验已有数据显示疗效并能预测其临床价值的;应对重大突发公共卫生事件急需的疫苗或者国家卫生健康委员会认定急需的其他疫苗,经评估获益大于风险的 药物临床试验期间 优先审评审批 临床急需的短缺药品、防治重大传染病和罕见病等疾病的创新药和改良型新药;符合儿童生理特征的儿童用药品新品种、剂型和规格;疾病预防、控制急需的疫苗和创新疫苗;纳入突破性治疗药物程序的药品;符合附条件批准的药品 上市许可申请前 特别审批 突发公共卫生事件时,国家药品监督管理局依法决定的应急所需防治药品 提出注册申请前 -
纳入到“突破性治疗”审评通道的药物,药审中心会优先处理有关沟通交流,加强指导并促进药物研发进程;在申报上市环节,该药物可纳入优先审评审批程序,审评时限缩短;上市申请阶段,药审中心会滚动接收其申报资料,并优先安排核查、检验等,这一系列措施可大大缩减新药从研发到上市的时间。2020−2022年间,共有12个药品通过突破性治疗程序上市(表2)。
表 2 2016−2022年我国加快审批途经的注册申请数及获批品种数
年份
(年)突破性治疗(件) 附条件批准的
品种数(个)优先审评 特别审批的
注册申请(件)纳入的注册申请 批准的新药上市申请 纳入的注册申请(件) 批准上市品种数(个) 2016 − − − 193 7 − 2017 − − − 230 50 − 2018 − − − 313 83 − 2019 − − − 253 82 − 2020 24 0 6 219 121 59 2021 53 5 38 115 131 81 2022 56 7 31 74 75 51 注:数据来源于NMPA官网。 -
“附条件批准”目的在于缩短药物临床试验的时间,使其尽早应用于无法继续等待的危重疾病或公共卫生方面急需的患者。符合附条件批准上市情形的药物,可使用替代终点、中间临床终点或早期临床试验数据来反映药物的有效性,当这些数据能够提示药品的获益大于风险时候,即可申请附条件批准上市。2020−2022年间,共有75个药品通过附条件批准程序上市(表2)。附条件批准可以有效缩短临床研发所需的时间,但也存在一定的风险。因对药品上市时临床安全性等要求的降低,增加了疗效不确定的可能性[5],存在安全有效性不足和资金浪费的风险,对于此类尚无充分证据的药品,平衡满足突发公共卫生事件药品需求和临床安全性可控,科学设计附条件批准方案,是监管部门面临的重要问题。
-
“优先审评审批程序”自2016年发布以来,在实践经验基础上不断优化调整,适用范围更多地向具有明显临床价值、临床急需的药物聚焦,致力于将更多的临床价值显著、临床急需的短缺药品、防治重大传染病、罕见病、儿童用药、纳入突破性治疗程序、符合附条件批准的药品等纳入优先审评程序。同时加速审评时限,药品上市许可申请的审评时限一般为200个工作日,优先审评审批程序的审评时限缩短至130个工作日,其中临床急需境外已上市罕见病用药优先审评审批程序的审评时限为70个工作日。至2022年,共有1 300余件注册申请被纳入该程序,其中获批上市品种数为549个(表2)。
-
“特别审批程序”其核心为出现突发公共卫生事件时,国家药品监督管理部门按照统一指挥、早期介入、快速高效、科学审批的原则,对应急处理所需药品进行特别审批,在申请受理、技术审评、抽样检验、行政审查等环节中优先处置,缩短审批时间。新冠肺炎疫情期间,特别审批程序在新冠病毒疫苗和治疗药物的审批中发挥了重大作用。2020年,共计59件与抗击新冠有关的中药、化学药、生物制品注册申请纳入该程序并完成技术审评,其中建议附条件批准上市1件,建议批准临床试验申请53件,增加适应证的补充申请5件。2021年与2022年分别审结81件及51件纳入特别审批程序的注册申请(均为新冠病毒疫苗和治疗药物),见表2。特别审批程序是一项制度性突破,但随着时间推移,该程序逐渐暴露出实施细节模糊、终止程序缺失等问题[6],不能适应当今国家公共卫生形势的新变化。
我国药品监管部门正在积极推进药品加快上市审评审批制度的改革,并形成初步的监管体制。2023年3月,药审中心总结抗疫应急审批经验,结合已有快速审批制度,制定了《药审中心加快创新药上市许可申请审评工作规范(试行)》[7]。随着改革的发展,更多深层次的问题也会随之暴露。为从根本上解决审评时限长、效率低等问题,药品监管部门应借鉴发达国家的一些加快审评审批政策,并结合中国国情构建一个具有中国特色的药品应急审评审批机制。
-
美国食品药品监督管理局(FDA)建立了4种药品应急审批的途径,包括快速通道(FT)、突破性疗法认证(BT)、优先审评(PR)及加速审批(AA)[8]。① FT:FT可以在药物研发的任何阶段由企业向FDA申请,主要针对在治疗严重的或危及生命的疾病方面具有一定潜力的新药审批。对进入FT的药物,FDA将进行早期介入,以使该药物“少走弯路”,加快研发进程。此外,申请人还可以在早期与FDA沟通,分阶段递交申报资料,不必一次性提交完整的申报资料,而FDA的审批将基于所治疗疾病的严重程度,按风险/效益原则进行评价。②BT:BT主要针对临床试验周期较长的药物,要求申请者提交早期临床试验数据,在药物临床试验阶段助力审批加速。③PR:PR在申请新药上市时提出,FDA会在60 d内做出是否批准优先审评的决定,主要针对与已上市药品比较有显著改进的药品上市申请,并不要求是具有全新分子实体的创新药,但临床疗效必须优于已上市药物。PR的时间为6个月,PR通道并不影响临床试验阶段的周期长短,也不降低审评标准。④AA:AA可使用替代终点、中间临床终点作为许可基础,允许在确切的治疗效果证据未全部收集到之前批准新药上市,主要针对治疗严重的、危及生命的疾病的新药,其批准一般附有条件,即药品具有可观察到的重大短期临床效果,而长期临床疗效则需进一步研究证明。这4种程序之间既存在差异又相互关联,同一个药品申请可适用多种应急审批途经。2018−2022年,FDA年均批准近50个新药,其中约68%的新药使用了一个或多个应急审批途径,具体见表3。
表 3 2018−2022年FDA通过加快审批途经批准的新药情况
年份
(年)批准新药总数(个) 加快审批途经批准的新药数量(个) 使用1个或多个加快途经的新药数量及
占批准总数的百分比[个(%)]快速通道 突破性治疗 优先审查 加速审批 2018 59 24 14 43 4 43(72.9) 2019 48 17 13 28 9 29(60.4) 2020 53 17 22 30 12 36(67.9) 2021 50 18 14 34 14 37(74.0) 2022 37 12 13 21 6 24(64.9) 注:数据来源于FDA官网。 此外,美国还建立了紧急使用授权(EUA)制度,即公共卫生处于紧急状态或存在陷入紧急状态的危险时,FDA可对未批准上市的医药产品或已获批准上市医药产品的其他用途进行授权[9]。同时,一旦官方宣布紧急情况结束时,所有基于该声明发布的EUA将不再有效。EUA的发布、更新和终止通知将在《联邦公报》(FR)上公布,并在FDA与美国疾病控制与预防中心(CDC)官网上公告。自2004年EUA制度建立以来,2009年之前FDA只发布过2个EUA。2009−2010年为应对H1N1流感,FDA为22个产品发布EUA,包括药品、诊断试剂和医疗器械。2012年以来,FDA先后对H7N9流感病毒、埃博拉病毒、寨卡病毒、COVID-19等发布多个EUA产品。截至2023年6月,FDA共批准了15个COVID-19治疗药物和4个疫苗的EUA[10]。
-
欧洲药品审评管理局(EMA)承担欧洲的药品审评审批工作,拥有来自欧盟各国的超过4 000多名专家组成的团队。EMA采取了多种新药应急审批的途径,包括附条件上市许可(CMA)、AA、特殊情况授权(EC)、优先药物审批(PRIME)等。
CMA主要针对未被满足的医疗需求,允许在临床数据不完整的情况下进行早期批准上市,上市后完成确证性临床试验。CMA的有效期为一年,可每年续签。上市许可持有人必须在规定的时间内履行特定义务,包括完成正在进行的或新的研究,或收集额外的数据。一旦上市许可持有人履行了所规定的义务,并且完整的数据证实该药物的益处继续大于其风险,上市许可就可以转换为标准上市许可(不再受特定义务的约束)。对于任何药物,如果新数据显示该药物的益处不再大于其风险,EMA可以暂停或撤销上市许可。2006−2016年间,EMA共有30个附条件上市药物获批,其中11个转为标准授权,2个因为商业原因撤回,其余17个仍处于附条件上市中[11]。
AA可将审批时间从标准程序的210 d缩短至150 d。申请人应证明其申报的医药产品预计具有重大公共卫生利益,特别是从治疗创新的角度来看。另外,申请人应提供有关GMP和GCP方面的信息,以便将常规GCP和批准前的GMP检查纳入加速评估程序。
EC允许患者获得无法根据标准授权批准的药物,在特殊情况下,EMA可能在没有全面数据的情况下授予上市许可。无法获得全面数据的原因在于只有极少数患者患有这种疾病,或是收集有关药物疗效和安全性的完整信息是不道德的。与附条件的上市许可不同,特殊情况授权可能在授权后也无法获得全面的数据。这些药物取得特定的授权后有义务接受EMA的监测约束。
此外,EMA还于2016年3月启动了PRIME计划,该计划目的在于加强对未满足医疗需求的药物,尤其是有潜力带来重大治疗成果的药物开发的支持。PRIME建立在现有的监管框架基础上,通过尽早与药物研发企业合作,为企业提供科学建议和加速评估,并确保患者只参与旨在生成必要数据的必要试验,从而充分利用有限的资源,使药物能够更早地到达患者手中。2016年1月至2021年6月,共有384个药物申请加入PRIME,其中95个被纳入该计划,年平均纳入率为25% [12]。2019−2022年EMA通过AA途经批准的新药情况见表4。
表 4 2019−2022年EMA通过加快审批途经批准的新药
年份
(年)批准新药
总数(个)加快审批途经批准的新药数量(个) 附条件
上市许可加速审批 优先药物
审批特殊情况
授权2019 66 8 3 0 1 2020 97 13 6 8 5 2021 92 13 3 6 4 2022 89 9 5 8 5 注:数据来源于EMA官网。 -
日本药品与医疗器械管理局(PMDA)负责新药审批,药品应急审批程序包括优先审评、先驱审查认定、附条件审批、再生医学产品有条件和有时限的批准、紧急授权、特例审批等。“优先审评”主要用于具有重大临床价值创新药物及孤儿药的注册审评,2021年,PMDA共批准了144个新药,其中56个通过优先审评,平均审批时间为223 d,同期标准审批时间为300 d。同时,PMDA为了减少不同审查员之间可能存在的人为因素干扰,制定了细致的审查标准化手册,改善了不同机构之间可能存在的信息传递滞后、人员沟通困难等问题,将药品审批时间大幅缩短。
“先驱审查认定”制度自2015年起开始试行,2020年正式提升到法规级别[13]。申请先驱审查的药品需满足治疗方法的突破性与革新性,针对重大严重影响生命质量的疾病或无法根治的疾病等要求。纳入先驱审查认定制度的药品享有优先咨询、加强预评估、优先审查等权力,该制度的审批时限为6个月,比以往减少一半。截至2023年6月,共有200余个药品申请加入先驱审查认定,其中25个药品被成功纳入,该25个药品中的17个已获批上市[14]。其余快速审批制度的对比见表5,通过快速审批,PMDA共批准了9个新冠治疗药,8个新冠疫苗产品(截至2022年末)[15]。
表 5 日本平时与紧急情况下的药品审批制度对比
对比项目 平时根据药品性质进行审批 紧急情况下的快速审批 附条件审批 再生医学产品有条件和
有时限的审批特例审批 紧急授权 对象 罕见病用药产品、开创性用药产品或特殊用途用药产品以及其他有特殊医疗需求的用药产品 非同源再生医学及其他产品(细胞/组织产品、基因产
品等)在外国(拥有与日本医药制度同等标准的制度的国家)销售的医药产品和其他产品 所有医药产品 制度宗旨 对医疗需求量大,但很难对足够数量的受试者进行临床试验以验证其疗效和安全性的医药产品给予批准 考虑到再生医学产品的特点(产品质量参差不齐,药理作用表现不一),对那些经少量病例证实安全且假定有效的产品予以批准 为了在紧急情况下防止健康危害的扩散,批准在外国销售的医药产品等 药品和其他产品的安全性已得到确认,其疗效也已得到推定,因此可获得批准,以防止紧急情况下健康危害的扩散 有效性 确认 推定 确认 推定 安全性 确认 确认 确认 确认 -
美国、欧盟、日本的药品优先审评目标定位明确,均以临床需求为目的,旨在加快临床急需药品的审评上市,形成覆盖全流程的多通道、多机制、多模式优先审评体系。制度中具有明确的适用范围、纳入标准、具体申请、受理、审核、审评流程及相关细则。法规体系层次清晰、完整,可操作性强。药审部门制定沟通方案和计划、提前沟通、主动指导。同时,为保证上市药品安全有效,各国针对通过优先审评上市的药品建立了更为严格的上市后监管制度,明确职能定位、监管范围、监管方式、申请人责任和义务,确保监管的科学、规范、有效。美国、日本在推进药品监管领域加快上市注册程序同时,还设置紧急授权使用药品作为应对公共卫生或突发事件的措施。以上这些,都对我国有很好的借鉴意义。
-
确定药品需求导向,进一步优化应急审批制度中的药品纳入范围,明确和细化纳入标准。建立符合我国国情的药品优先审评上市后的监管制度,对药品上市后研究、限制性适用、信息公开、撤销上市或转为普通上市,以及相关的强制性手段、法律责任等都应予以明确细化的规定[16]。
建立我国药品EUA制度。我国《中华人民共和国突发事件应对法》对突发事件的预防和应急准备等作出了明确规定,但目前仍缺少关于药品紧急使用授权的法律依据。建议建立我国药品EUA制度,组建由国家卫生健康委员会、药品监管部门及相关部门组成的EUA专业领导工作组协调、管理、实施EUA。应急情况下可考虑对未经药品监管部门批准上市的药品,在具有基础研发数据,可以证明其安全性和有效性的前提下,启动EUA。EUA是在公共健康利益受到严重威胁和药品安全有效评价之间寻找的平衡,遵循“边审批、边使用、边评估、边调整”的原则,可以与加快注册评审同步进行,及时补充调整新药的使用限制。同时,出台EUA实施细则,明确规定实施EUA的条件、发布程序、审评流程和要求、终止或撤销、产品覆盖范围、信息公开、告知义务、责任保护等[17]。
-
目前的药品应急审批程序主要从促进申请人与药审中心沟通、调整上市标准这两方面来提高新药注册的效率。我国虽设置了药品审评专家咨询委员会,但人员配置数量较欧盟、日本等较少,在药品审评过程中需要召开会议解决问题,或是申请过程中存在争议主动征询意见时提供相应的技术指导与决策建议,发挥作用有限。应积极推动审评审批人才培养和队伍建设,扩大药审专家职能,实现人员配置的优化和审评通过率的提升。
药品的上市审批过程中,申请方与监管部门、检验机构之间存在信息交流滞后的问题,应尽快推广和加强药审中心受理人员及项目管理人员的提前介入,争取受理问题在申报前得到解决,降低企业的创新风险,同时防止审评资源的浪费与人力、物力的消耗。
-
美国、欧盟、日本药品优先审评制度中,药审部门侧重沟通指导,并重视上市后药品的安全性与有效性,建立了严格的上市后监管制度。现阶段,我国加快上市注册程序对新药的开发促进作用较为明显,通过快速审批程序注册上市的药品逐年增多,为避免相关药物上市后出现无期望的临床疗效或存在安全性问题,急需加强快速审批药品上市后的监管工作,保证审批药物的安全有效。新版《药品注册管理办法》引入药品“加快上市注册程序”的同时也明确设立了上市退出机制。为实现此类特殊审评通道药品上市前、后安全监管工作的顺利衔接,可借鉴EMA的经验,细化特殊审评药品准入条件,将上市前风险管理体系的建立作为关键审评因素,上市后设置额外监测、风险沟通等安全监管制度进行有效衔接,从而实现对用药风险的持续监测和动态管理。
-
随着公众对药品上市的效率以及对突发事件应急药品供应的需求提升,我国药监部门也尽力在保障药品安全有效的前提下,出台一系列政策以尽量缩短紧急必需用药的审评审批时间。在应对新冠疫情过程中,药品应急审批工作已取得了诸多进展。然而,对比国外药品快速审批程序,我国目前仍处于发展阶段,因此需认真总结既往的工作经验,并在实践过程中不断完善。同时借鉴一些国外先进模式和监管方式,进一步改善我国药品应急审批制度体系和流程,提升药品审批效率,提高应对突发事件的药品保障能力。
Comparative study on pharmaceutical emergency approval systems in China and other countries
-
摘要:
目的 通过对比中外药品应急审批制度,探讨进一步优化我国药品应急审批制度的思路和措施。 方法 分析我国药品应急审批制度现状,并与美国、欧盟、日本的相关制度对比。 结果 国外药品应急审批目标定位明确,以临床需求为目的,加快临床急需药品的审评上市,形成覆盖全流程的多通道、多模式、多机制优先审评体系,法规体系层次清晰、完整,可操作性强。我国相应的审批制度起步较晚,处于不断完善的阶段,在制度建设、政府职能、人员优化、上市后监管等方面有待改进。 结论 我国药监部门应总结既往工作经验,借鉴国外一些可行的审批模式和监管方式,进一步改善药品应急审批制度体系与流程,提升药品审批效率以及应对突发事件的药品保障能力。 Abstract:Objective To explore the ideas and measures to further optimize China’s drug emergency approval system by comparing the drug emergency approval systems in China and other countries. Methods The current situation of China’s drug emergency approval system was analyzed and compared with the relevant systems of the United States, the European Union and Japan. Results The goal of drug emergency approval in other countries is clearly positioned to accelerate the review and listing of urgently needed clinical drugs for the purpose of clinical needs, forming a multi-channel, multi-mode and multi-mechanism priority review system covering the whole process, and the regulatory system is clear, complete, and operable. The corresponding approval system in China started late and is in the stage of continuous improvement, which needs to be improved in terms of system construction, government functions, personnel optimization, and post-market supervision. Conclusion Drug regulatory authorities in China should summarize their past work experience and draw on some feasible approval models and regulatory approaches in foreign countries, further improve the system and process of the drug emergency approval system and enhance the efficiency of drug approval and the ability to protect drugs in response to emergencies. -
Key words:
- drugs /
- emergency approval /
- institutional comparison /
- emergency authorization
-
粉-液双室袋是采用特定工艺将药物和注射用溶剂独立封装在不同的两个腔室中的一种静脉注射用产品,在医护人员紧缺或战备、紧急救援等情况下,其优势突出[1]。1996年,日本研制出世界首个粉-液双室袋产品——头孢唑林钠氯化钠注射剂[2]。2015年,原中国食品药品监督管理总局出台首个《粉液双室袋产品技术审评要点》[3],国内首个粉-液双室袋产品(注射用头孢他啶/氯化钠注射液)于2019年获得药品注册批件,正式上市。粉-液双室袋因其结构上的创新,给药预处理步骤简化在使用上具有独特的优势,因此粉-液双室袋产品自上市后就受到了广泛的关注。但对于这样一个新产品,是否真正安全有效,是否具有成本-效益,与市场上正在使用的传统粉针剂相比是否具有明显的优势等问题目前均未得到解答,利益各方大都采取观望的态度。
为促进粉-液双室袋产品临床合理应用,依据国家卫健委2020年发布的《药品临床综合评价管理指南(试行)》[4],通过对文献资料进行调研,提取粉-液双室袋常用评价指标,以传统粉针产品为对照,从安全性、有效性、经济性、适宜性、可及性、创新性6个维度对粉-液双室袋产品进行综合评价。
1. 资料和方法
1.1 文献检索策略
以“双室袋” “双腔袋” “多室袋” “多腔袋” “Multi chamber bag”和“dual chamber bag”等为关键词,在中国知网、万方数据、维普、PubMed 、Web of Science等数据库中进行系统文献检索,对发表年度不设限制。表1为具体的检索式及相应检索结果。
表 1 检索式及检索结果检索条件 各数据库检索结果 中国知网 万方数据 维普 PubMed Web of Science (主题=双室袋 + 粉液双室袋)OR(主题=双腔袋)
OR(主题=多室袋)OR(主题=多腔袋)80 ((((任意字段=双室袋 OR 任意字段=粉液双室袋)OR 任意字段=多室袋)OR 任意字段=双腔袋)OR 任意字段=多腔袋) 189 主题=(双室袋)OR 主题=(粉液双室袋)OR 主题=(多室袋)
OR 主题=(双腔袋)OR 主题=(多腔袋)6740 (Multi chamber bag)OR(dual chamber bag) 42 (Multi chamber bag)OR (dual chamber bag) 166 1.2 纳入与排除标准
纳入标准:①研究对象为粉-液双室袋;②内容为安全性、有效性、经济性、适用性、创新性、可及性的研究;③文献类型为随机对照试验或观察实验;④有参考价值的多室袋研究文献。
排除标准:①研究对象为液-液双室袋;②内容为生产工艺、设备、分装技术和其他无关内容;③文献类型为综述、会议论文、专利、成果;④重复文献、不可下载文献。
1.3 文献筛选和信息提取
文献由2名研究员独立、同步进行筛选。参考中国医药包装协会发布的《基础输液临床使用评估指南(试行)》[5]和国家卫健委发布的《药品临床综合评价管理指南(试行)》[4]提取文献中的可用指标。如产生争议,由课题组成员讨论决定。
2. 结果
2.1 文献检索与筛选结果
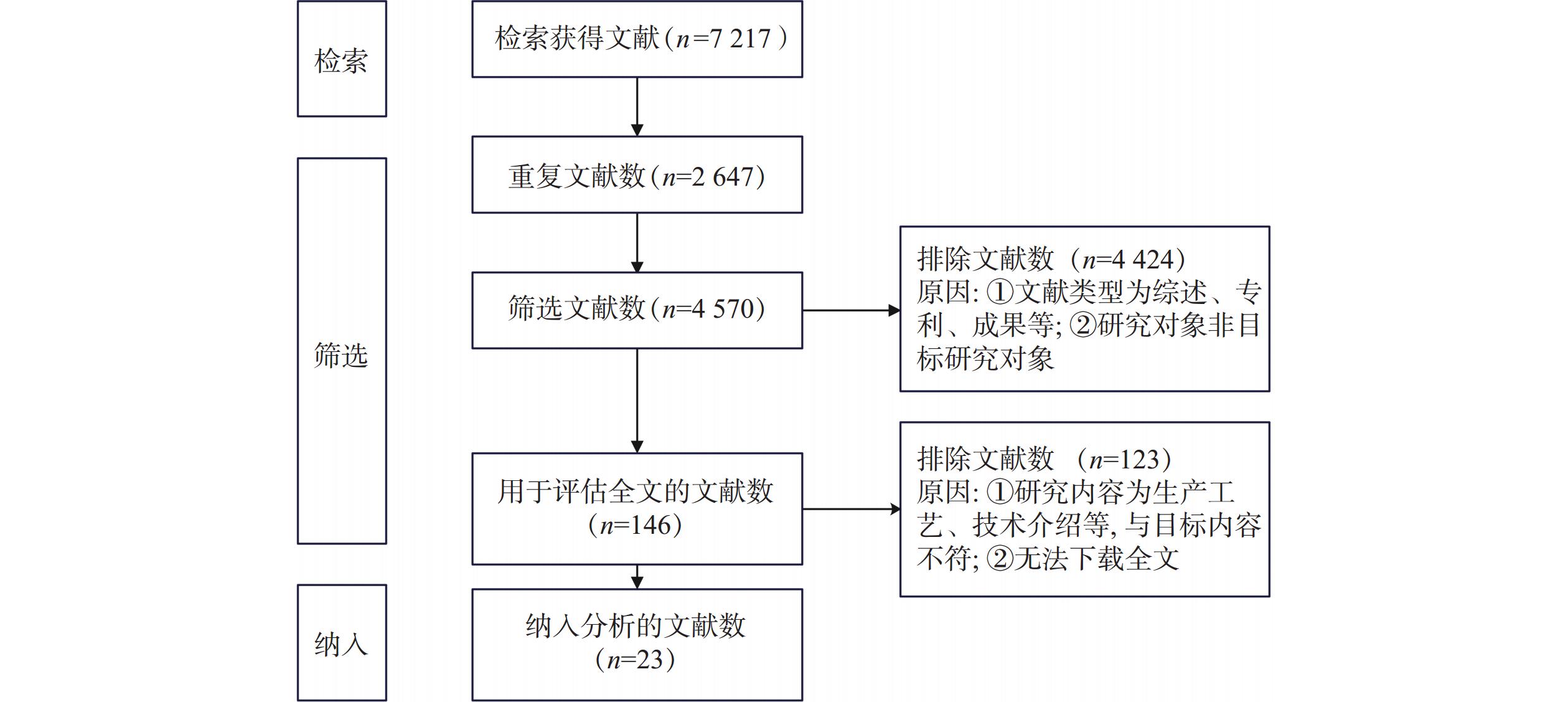
系统检索文献得
7217 篇,排除重复文献2647 篇、文献类型为综述、会议论文等4424 篇、文献内容为生产工艺和技术介绍等123篇,最终纳入分析文献23篇,其中英文文献2篇,中文文献21篇。具体筛选过程如图1。2.2 指标提取及评估结果
2.2.1 评价指标
文献中使用的评价指标涉及5个维度,经过整合、汇总,见表2。由于双室袋产品和传统粉针剂的药物成分、给药途径一致,仅包装和给药预处理存在区别,故有效性、安全性两个维度仅包括与包装和给药预处理相关的指标。其中,虽配制过程刺伤、划伤等为操作失误,但双室袋产品简化了药液配制过程,无需使用注射器辅助配制,可完全避免意外伤害,与传统粉针剂存在差异,故纳入为安全性指标。
表 2 已发表文献使用的评价指标评价维度 判别可用指标 有效性 药液稳定性、配制浓度准确性、药液残留量 安全性 不溶性微粒、刺伤划伤等意外事情发生率 经济性 配制成本a、废弃物重量、住院成本、血液感染发生率 适宜性 配制时间、平均医护人员人力占用、包装重量和
储运体积b、环境适应性、废弃物处理难易程度可及性 生产厂家数量、产品原材料供应能力、患者可负担性 a:为药液成本、配制用品以及配制人工成本的总和;b:包括药液配制过程所需用品储运体积总和,传统粉针剂产品的配制用品包括注射器、西林瓶粉针、配制用溶剂等,粉-液双室袋产品仅包括产品本身。 2.2.2 有效性
于庆坤等[6-12]研究结果显示,与传统粉针剂相比,双室袋产品的药液含量随时间变化小,配制的实际浓度更接近理论浓度,无残留药液,具体见表3和表4。
表 3 药液配置后5 h的稳定性对比[7]产品名称 不同温度时的百分含量(%) 4℃ 25℃ 非PVC粉-液双室袋产品 96.33 96.32 玻璃瓶粉针产品 95.37 95.79 表 4 配制浓度准确性以及药液残留量的比较2.2.3 安全性
静脉输液中的不溶性微粒会造成血栓和静脉炎[13],药典规定用于静脉注射、滴注的药品需检查不溶性微粒。双室袋法配制过程不溶性微粒无明显增加[8,10,14-15],详见表5。以18名护士为观察对象,粉针配制过程1名护士被划伤,双室袋配制过程无人员刺伤、划伤[10]。粉-液双室袋以非PVC多层共挤膜为膜材,其强光照射实验表明0.9%氯化钠、5%葡萄糖、葡萄糖氯化钠、复方氯化钠注射液与膜材相容性良好,稳定性试验表明非PVC多层共挤膜的水蒸气渗透、透光率、pH、易氧化物检测均符合规定[14,16]。
表 5 配制过程不溶性微粒比较文献作者 样品 不溶性微粒数(个/ml) ≥5 ≥8 ≥10 ≥12 ≥25 ≥100 李英等[15]a 粉针输液产品 318 66 21 6 0 0 配制增加微粒数b 285 55 15 3 0 0 双室袋输液产品 2 0 0 0 0 0 双室袋增加微粒数c 0 0 0 0 0 0 沈敏娜等[16]a 粉针输液产品 322 68 23 7 0 0 配制增加微粒数 289 55 17 5 0 0 双室袋输液产品 3 1 0 0 0 0 双室袋增加微粒数c 0 0 0 0 0 0 王宇航等[8]a 粉针输液产品 240 326.5 43 24 7 0 双室袋输液产品 240 2 0 0 0 0 罗莉等[10]d 粉针输液产品 219.52±84.73 43.93±21.68 14.93±7.96 4.05±2.60 0.01±0.04 双室袋输液产品 3.49±0.95 0.39±0.19 0.20±0.11 0.13±0.09 0.03±0.03 a:实验重复配制(均≥100份),由于数据资料不服从正态分布,选中位数表征平均水平,表中均为中位数值;b:增加微粒数=溶液微粒数−(粉体+液体),表示溶配方法增加的不溶性微粒;c:双室袋法配置的不溶性微粒增加数计算结果为负值,由于混合前为取出粉末后在非封闭的环境中进行溶解测试,而混合后则是在封闭的袋内进行开通溶解后测试,从而导致粉体检测结果高于混合液的情况出现,因此即配型双室袋法配置的不溶性微粒增加数视为零;d:本实验所取数据为不溶性微粒数范围值。 2.2.4 经济性
在纳入的双室袋产品相关经济性研究[8,17-18]中考虑到的成本指标包括配制成本(包括配制环境、设备以及环境维护费、人工成本、耗材成本)、废弃物重量,效果/效益以血液感染(BSI)为指标。采用双室袋输液,药液配制过程得到简化,一方面可节省储存场所、人工及耗材成本,一方面产生的废弃物减少,废弃物处理费会有所减少。王宇航等[8]通过计算发现按照4.82元/kg的垃圾清理费算,每年可减少废弃物处理费约7万元。此外,双室袋输液配制过程减少与空气的接触,可在一定程度上减少输液后血液感染的发生率。苗雅楠等[17]发现在
1000 例患者中,输液系统由半开放式转换为全密闭输液系统可减少172例血液感染。已上市的粉-液双室袋产品与对应的传统粉针相比,有效期相同,对于储备来说,两者轮换周期一致。2.2.5 适宜性
对于易溶于水但在水溶液中不稳定的药物一般选择制成注射用无菌粉针,此类产品在使用前需要与特定的配制用溶剂临时配制注射用溶液。与粉针剂西林瓶产品运用的药液配制方法相比,双室袋法输液预处理环节有所简化,能明显缩短药液配制时间[8,10,19],具体见表6,在医护资源匮乏(如应急医学救援)时,可减少医护人员人力的占用。此外双室袋为全密闭输液系统,对环境的耐受性强。
2.2.6 可及性
可及性包括可获得和可负担两方面的要求。双室袋产品的药液与传统粉针一致,包装材料有所不同,目前双室袋包材审批通过已登记的有15项(1项为进口),双室袋产品生产厂家超过5家,产品大都为头孢类抗感染药物,在生产供应方面,可以满足可及性的要求。此外,2022年《国家基本医疗保险药品目录》的协议期内谈判药品部分有5个粉-液双室输液产品被纳入,为医保乙类药品,可达到患者可负担的要求。
2.2.7 创新性
截止到2023年4月,与粉-液双室输液袋有关的发明专利有22项,实用新型专利89项,外观专利5项,包括生产、灌装、检漏等方面。粉-液双室袋产品能缩短配液时间,简化输液预处理过程,降低了输液对环境的要求,顺应了突发事件应急医学救援与创伤急救的需求。
3. 讨论
本研究采用系统综述的方法对现有文献信息分析提取,得到的指标可作为评价指标池的一部分,后续可用于建立粉-液双室袋产品综合评价指标体系。
粉-液双室袋产品与传统粉针药效成分、给药途径相同,我国对药品上市后包装变更无临床试验的要求[20],已发表文献中均未报告其有效性、安全性的临床结局指标,且两者仅在包材和给药预处理方面存在差异,故纳入的有效性指标只包括药物学工艺重现性,安全性指标只包括不良事件或风险的比较指标。除列出指标外,安全性评价还需考虑渗漏隐患、破损率、误配、错配发生率等。经济性评价具有时间性,存在偏倚风险,仍需对此类产品进行更为可靠的经济学研究。适用性的考查应注重产品在紧急救援使用时缩短抢救时间,节约医护资源的能力。粉-液双室袋的技术壁垒较高,可及性方面不如传统粉针剂,但符合可及性的基本要求。
粉-液双室袋属于全封闭式输液系统,无空气通路,细菌污染降低,减少了输液反应的发生[21]。药液配制时间明显缩短,提高救援成功率[22];无需临时计算溶剂用量,配制准确度高,无药液残留,且对环境、技术要求不高,降低了人工、设施及耗材成本;无需借助注射器反复穿刺橡胶塞,不溶性微粒显著减少,刺伤、划伤等意外事情可避免。虽单价高于传统粉针产品,但其在废弃物处理成本、人工成本以及输液后静脉炎发生率的减少方面展现出了一定的优势。此外国内双室袋产品的生产企业超过5家,且在原材料上摆脱了对进口的依赖,5种粉-液双室袋产品已纳入医保目录,符合可及性要求。
粉-液双室袋为新兴产品,仅有23篇符合纳入条件。外文文献较少,仅纳入2篇。因多室袋与双室袋原理一致,故将以多室袋为研究对象的文献也纳入。期待随着粉-液双室袋在我国广泛应用,未来可获得高质量、精细化的研究数据,以开展更全面、可靠的综合评价研究。
-
表 1 我国药品应急审批程序对比
名称 适用范围 申请阶段 突破性治疗药物 在药物临床试验期间,用于防治严重危及生命或严重影响生存质量的疾病且尚无有效防治手段或与现有治疗手段相比具有明显临床优势的创新药或改良新药等 在Ⅰ、Ⅱ期临床试验阶段,通常不晚于Ⅲ期临床试验开展前 附条件批准 治疗严重危及生命且尚无有效治疗手段的疾病的药品,药物临床试验已有数据证实疗效并能预测其临床价值的;公共卫生方面急需的药品,药物临床试验已有数据显示疗效并能预测其临床价值的;应对重大突发公共卫生事件急需的疫苗或者国家卫生健康委员会认定急需的其他疫苗,经评估获益大于风险的 药物临床试验期间 优先审评审批 临床急需的短缺药品、防治重大传染病和罕见病等疾病的创新药和改良型新药;符合儿童生理特征的儿童用药品新品种、剂型和规格;疾病预防、控制急需的疫苗和创新疫苗;纳入突破性治疗药物程序的药品;符合附条件批准的药品 上市许可申请前 特别审批 突发公共卫生事件时,国家药品监督管理局依法决定的应急所需防治药品 提出注册申请前  下载: 导出CSV
下载: 导出CSV
表 2 2016−2022年我国加快审批途经的注册申请数及获批品种数
年份
(年)突破性治疗(件) 附条件批准的
品种数(个)优先审评 特别审批的
注册申请(件)纳入的注册申请 批准的新药上市申请 纳入的注册申请(件) 批准上市品种数(个) 2016 − − − 193 7 − 2017 − − − 230 50 − 2018 − − − 313 83 − 2019 − − − 253 82 − 2020 24 0 6 219 121 59 2021 53 5 38 115 131 81 2022 56 7 31 74 75 51 注:数据来源于NMPA官网。
下载: 导出CSV
表 3 2018−2022年FDA通过加快审批途经批准的新药情况
年份
(年)批准新药总数(个) 加快审批途经批准的新药数量(个) 使用1个或多个加快途经的新药数量及
占批准总数的百分比[个(%)]快速通道 突破性治疗 优先审查 加速审批 2018 59 24 14 43 4 43(72.9) 2019 48 17 13 28 9 29(60.4) 2020 53 17 22 30 12 36(67.9) 2021 50 18 14 34 14 37(74.0) 2022 37 12 13 21 6 24(64.9) 注:数据来源于FDA官网。
下载: 导出CSV
表 4 2019−2022年EMA通过加快审批途经批准的新药
年份
(年)批准新药
总数(个)加快审批途经批准的新药数量(个) 附条件
上市许可加速审批 优先药物
审批特殊情况
授权2019 66 8 3 0 1 2020 97 13 6 8 5 2021 92 13 3 6 4 2022 89 9 5 8 5 注:数据来源于EMA官网。
下载: 导出CSV
表 5 日本平时与紧急情况下的药品审批制度对比
对比项目 平时根据药品性质进行审批 紧急情况下的快速审批 附条件审批 再生医学产品有条件和
有时限的审批特例审批 紧急授权 对象 罕见病用药产品、开创性用药产品或特殊用途用药产品以及其他有特殊医疗需求的用药产品 非同源再生医学及其他产品(细胞/组织产品、基因产
品等)在外国(拥有与日本医药制度同等标准的制度的国家)销售的医药产品和其他产品 所有医药产品 制度宗旨 对医疗需求量大,但很难对足够数量的受试者进行临床试验以验证其疗效和安全性的医药产品给予批准 考虑到再生医学产品的特点(产品质量参差不齐,药理作用表现不一),对那些经少量病例证实安全且假定有效的产品予以批准 为了在紧急情况下防止健康危害的扩散,批准在外国销售的医药产品等 药品和其他产品的安全性已得到确认,其疗效也已得到推定,因此可获得批准,以防止紧急情况下健康危害的扩散 有效性 确认 推定 确认 推定 安全性 确认 确认 确认 确认
下载: 导出CSV
-
[1] 国家药品监督管理局. 药品特别审批程序[EB/OL]. (2005-11-18)[2023-10-11]. https://www.nmpa.gov.cn/yaopin/ypfgwj/ypfgbmgzh/20051118010101724.html. [2] 国家药品监督管理局. 总局关于解决药品注册申请积压实行优先审评审批的意见[EB/OL]. (2016-02-26)[2023-10-11]. https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20160226085101295.html. [3] 国家药品监督管理局. 国家药监局关于发布《突破性治疗药物审评工作程序(试行)》等三个文件的公告(2020年第82号)[EB/OL]. (2020-07-08)[2023-10-11]. https://www.nmpa.gov.cn/xxgk/fgwj/ xzhgfxwj/20200708151701834.html. [4] 国家市场监督管理总局.药品注册管理办法[EB/OL]. (2020-01-22)[2023-10-11]. https://www.gov.cn/zhengce/zhengceku/2020-04/01/content_5498012.htm. [5] 吴忠虹, 董丽. 我国药品优先审评审批制度的现状分析[J]. 中国处方药, 2022, 20(10):29-31. [6] 陈先红, 王闻雅. 新形势下我国药品特别审批制度的思考[J]. 中国食品药品监管, 2020(9):30-35. [7] 国家药品监督管理局药品审评中心. 《药审中心加快创新药上市许可申请审评工作规范(试行)》[EB/OL]. (2023-03-31)[2023-10-11]. https://www.cde.org.cn/main/news/viewInfoCommon/ace377c025ad4f2bbf 94790673b2646e. [8] FDA. Guidance for Industry: Expedited Programs for Serious Conditions–Drugs and Biologics[EB/OL]. (2014-05)[2023-10-11]. https://www.fda.gov/media/119748/download. [9] FDA. Emergency Use Authorization[EB/OL]. (2017-01-13)[2023-10-11]. https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization. [10] FDA. Emergency Use Authorization--Archived Information[EB/OL]. (2022-01-24)[2023-10-11]. https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization-archived-information. [11] EMA. Conditional marketing authorisation–Report on ten years of experience at the European Medicines Agency[EB/OL]. (2017-01-23)[2023-10-11]. https://www.ema.europa.eu/system/files/documents/report/wc500219991_en.pdf. [12] EMA. PRIME: Analysis of the first 5 years’ experience[EB/OL]. (2022-08-05)[2023-10-11]. https://www.ema.europa.eu/system/files/documents/report/2022-03_prime_5_years_report_updated_2022-04-05-en.pdf. [13] PDMA. 先驱医药品审查认定制度[EB/OL]. (2020-12)[2023-10-11].https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/kenkou_iryou/iyakuhin/tp150514-01_00001.html. [14] PDMA. 医药品先驱审查认定制度的对象品目一览表[EB/OL]. (2023-06)[2023-10-11]. https://www.pmda.go.jp/files/000235895.pdf. [15] PDMA. PMDA最新动向[EB/OL]. (2022-12-08)[2023-10-11].https://www.pmda.go.jp/files/000249287.pdf. [16] 冯媛媛, 杨悦. 我国药品优先审评制度的实施情况及建议[J]. 中国药房, 2018, 29(15):2026-2031. doi: 10.6039/j.issn.1001-0408.2018.15.03 [17] 王颖, 张小平, 杨依晗, 等. 突发公共卫生事件下的药械紧急使用授权政策思考[J]. 中国医药导刊, 2020, 22(3):157-161. -

