


 下载:
下载:


-
WHO提出抗菌药物耐药性是全球十大健康威胁之一[1],将耐碳青霉烯类肠杆菌(CRE)、耐碳青霉烯铜绿假单胞菌(CRPA)和耐碳青霉烯鲍曼不动杆菌(CRAB)列为最紧迫的威胁。美国疾病控制和预防中心(CDC)将CRE定义为对任何碳青霉烯类抗菌药物不敏感的肠杆菌(即对多利培南、美罗培南或亚胺培南的最低抑制浓度≥4 μg/ml或对厄他培南的最低抑制浓度≥2 μg/ml)或被证明会产生碳青霉烯酶的肠杆菌。碳青霉烯类抗菌药物是一类具有广谱抗菌作用的β-内酰胺类药物,通过抑制青霉素结合蛋白(PBPs)阻碍细菌细胞壁合成[2],曾被许多医院认为是抗菌治疗中的“最后手段”。 目前CRE分离株对大多数临床应用的抗菌药物具有耐药性,从而使临床中抗菌药物的选择受到限制[3]。据统计美国每年有超过
13000 例CRE感染患者,导致超过1000 人死亡[4]。2011年美国CRE感染的发生率为4.2%[5]。2010~2013年欧洲CRE感染的发生率为2.0%[6]。根据抗菌药物耐药性监测网(CHINET)显示,2014年我国CRE感染的发生率为12.5%,2016年为22.9%,2019年则升至26.8%。与2015年相比,2023年中国的CRE发生率增加了58%以上,因此,对CRE的耐药机制的研究值得我们重视。 -
碳青霉烯酶是一个多样化的β-内酰胺酶家族,具有水解和灭活多种抗菌药物的能力,包括青霉素类、头孢菌素类、单环β-内酰胺类和碳青霉烯类。这些酶与药物结合后破坏四元氮杂二酮环的酰胺键,从而阻止药物与细菌细胞壁青霉素结合蛋白结合[7]。使用Ambler分类系统,碳青霉烯酶可分为A类、B类和D类β-内酰胺酶。
A类碳青霉烯酶利用丝氨酸残基水解β-内酰胺结构[8],包括含blaKPC、blaNMC/blaIMI和blaSME基因的3种类型[9],其中,最常见的是含blaKPC基因[10],主要存在于肺炎克雷伯菌中,但也存在于多种其他肠杆菌中,包括弗氏柠檬酸杆菌、阴沟肠杆菌、大肠杆菌、粘质沙雷菌以及假单胞菌等 [9]。
B类金属β-内酰胺酶(MBLs)是锌依赖性的[11],包括含blaVIM、blaIMP和blaNDM基因的三种类型[11-13],它们都存在于可移动的遗传基因上,能够水平传播。金属酶类的β-内酰胺酶能够水解多种β-内酰胺类抗菌药物,但不能水解单环β-内酰胺类抗菌药物,如氨曲南。
D类碳青霉烯酶包括OXA编码基因的成员,主要存在于不动杆菌中,其中blaOXA-48型也存在于肠杆菌中[14-15],与CRE院内感染相关[16-18]。blaOXA-48型包括OXA-48及其相关变体OXA-181、OXA-162和OXA-232型等。
在中国,存在blaKPC和blaNDM基因是大多数CRE菌株表型耐药的原因[19-20]。其中肺炎克雷伯菌中产生KPC酶最多,其次为OXA-48酶(37%,310/850)和NDM酶(11%,93/850)。在大肠杆菌中,OXA-48酶(56%,43/77)最为常见,其次为NDM酶(26%,20/77)和KPC酶(18%,14/77)[21]。
-
外排泵是一种主动转运蛋白,主动从细菌中排出抗菌药物,减少了细菌中的药物浓度,使细菌产生耐药性。外排泵基因包括5个家族(SMR、MFS、RND、ABC和MATE家族),其中RND家族临床意义更为重要,它们能识别广泛的底物,与多药耐药相关[22]。最具特征的RND系统是AcrAB-ToIC,由膜融合蛋白AcrA(膜间质蛋白、周浆蛋白)、外排转运蛋白AcrB(内膜蛋白)以及外膜通道蛋白TolC(外膜蛋白)组成的三联体复合物,在肠杆菌中占有绝对优势[23-24]。AcrAB-TolC的调节分为局部抑制和转录因子的全局调控。局部抑制因子AcrR可作为调节剂阻止AcrAB的过表达,AcrR的突变使其抑制作用丧失,可导致AcrAB-TolC的过表达而产生多药耐药[25-26]。其他局部抑制因子还有AcrS/EnvR、组蛋白样核结构蛋白(H-NS)和分裂抑制因子(Sdi)A,但相对影响较小[27]。大肠杆菌中,基因表达主要由多重耐药操纵子MarA控制,在肠沙门氏菌中主要由RamA控制。大肠杆菌中的MarR、MarA和MarB基因参与多种抗菌药物耐药。MarR是一种在没有任何环境信号的情况下阻断自身转录的蛋白质[4],只有当MarR的抑制被破坏时,MarRAB的转录才会发生[28]。MarA的表达导致其调控中的许多基因被激活,包括AcrAB和TolC,从而增加药物外排和多重耐药。此外,氧化应激反应的发生也可能诱导AcrAB,从而产生耐药。
-
外膜孔蛋白(OMP)的缺乏或突变是碳青霉烯类耐药的潜在重要机制,也是耐碳青霉烯肺炎克雷伯菌、耐碳青霉烯鲍曼不动杆菌等耐碳青霉烯类肠杆菌的重要耐药机制。OmpK35和OmpK36是两种非特异性孔蛋白,抗菌药物及亲水小分子可通过这两种孔蛋白以被动扩散方式进入细菌内而发挥抗菌作用。编码序列或启动子位点的突变、插入、删除或替换会使孔蛋白的翻译过早终止,影响孔蛋白表达从而导致抗菌药物难以进入细菌,产生耐药性。另一种机制是在转录后水平上通过相对较小的sRNAs调节孔蛋白的表达导致耐药,有报道这种现象出现在刺激应激(包括氧化应激)和渗透压、pH值或温度改变下的大肠杆菌分离株与产气肠膜杆菌中[28]。部分研究表明Omp36比Omp35在碳青霉烯耐药中发挥更重要的作用[29-30],它们的缺失或缺乏与AmpC酶的结合可能导致高水平的碳青霉烯类耐药。
-
青霉素结合蛋白(PBPs)是存在于几乎所有细菌细胞内膜上的一类酶。它们的主要功能是合成细菌细胞壁肽聚糖,形成细胞外骨架。β-内酰胺类抗菌药物的靶蛋白可与细胞内膜上的PBP靶点特异性结合,从而干扰细胞壁肽聚糖的形成,最终导致细菌溶解[31]。PBPs的表达减少或突变可导致抗菌药物MIC值升高,PBPs的突变还可能在导致临床耐药的同时导致孔蛋白产量减少或碳青霉烯酶产生增加,从而发生细菌耐药[32]。
-
氨曲南是单环β-内酰胺类抗菌药物,对产生A类碳青霉烯酶的细菌没有活性,包括高度流行的产KPC碳青霉烯酶的细菌[33]。氨曲南对产生B类和D类碳青霉烯酶的细菌有效,但这些细菌通常携带伴随ESBL基因,可水解氨曲南使其无效,因此作为单一疗法,氨曲南的临床应用通常受到限制[33-34]。氨曲南与新型β-内酰胺酶抑制剂的复合制剂(头孢他啶-阿维巴坦)合用,是一种很有前途的治疗产金属β-内酰胺酶耐药菌的治疗方案。头孢他啶-阿维巴坦单独对金属β-内酰胺酶没有活性,氨曲南对B类碳青霉烯酶有活性但易被其他β-内酰胺酶水解,因此体外,头孢他啶-阿维巴坦和氨曲南之间存在显著的协同作用,可对产金属β-内酰胺酶耐药菌属产生抗菌活性[35]。
-
磷霉素依靠硝酸甘油-3-磷酸转运子(GlpT)和葡萄糖-6-磷酸转运体(UhpT)两种膜转运系统进入细菌内,可同时激活两种转运体使其转运更多磷霉素进入细菌[36]。磷霉素进入细菌后不可逆地竞争性结合丙酮酸-二磷酸尿嘧啶乙酰葡糖胺转移酶(UDP-NrA),参与肽聚糖生物合成的初始步骤[37-39]。肠杆菌科细菌对于磷霉素产生的耐药主要为获得性耐药,即当磷霉素进入细菌的通路发生改变时,细菌就会产生耐药性。例如,大肠埃希菌中编码磷霉素转运体的GlpT、UlpT或UhpT,以及转录上游调控基因UhpA突变都可导致磷霉素进入细菌减少;同时,GlpT和UlpT的表达依赖环磷酸腺苷(cAMP),cAMP表达调控基因ptsl和cyaA的突变导致胞内cAMP浓度降低,从而诱发GlpT和UlpT表达降低,磷霉素进入细菌减少。另外,编码磷霉素水解酶fosA基因也可直接修饰介导肠杆菌对磷霉素的耐药[40-41]。
-
多粘菌素类抗菌药物长期以来一直用于耐药革兰氏阴性菌抗感染治疗,被认为是治疗多重耐药微生物引起感染的一线治疗方案和最后的治疗选择之一,特别是对碳青霉烯类耐药的革兰氏阴性杆菌,其中包括CRE。然而多粘菌素的耐药性正在出现,染色体中点位突变导致细菌脂多糖膜改变或外排泵增加,或由质粒介导的MCR基因改变脂多糖膜中的脂质A导致无法与多粘菌素结合,都会产生耐药性[42-43]。因为多粘菌素具有显著的肾毒性,且通常对携带A类碳青霉烯酶的分离株疗效较低,所以美国感染病学会(IDSA)2023版指南目前不推荐多粘菌素用于治疗CRE。尽管新的抗感染药物已被批准用于临床使用,但世界许多地区都无法获得新药,此时多粘菌素往往还是CRE感染唯一可用的抗菌药物[44-45],因此,其仍被世卫组织认为是“最优先”的至关重要的抗菌药物。
-
替加环素是在米诺环素核心D环的C9碳处添加了N,N-二甲基甘氨酰胺。因此替加环素有不受四环素类抗菌药物主要耐药机制影响的特性,如四环素特异性外排泵和核糖体保护机制。替加环素通过可逆结合细菌核糖体30S亚基上的螺旋区(H34)来抑制细菌蛋白翻译。随着替加环素在耐碳青霉烯肺炎克雷伯菌感染临床治疗中应用广泛,对肠杆菌科细菌耐药的报道也逐渐增多[46]。替加环素耐药机制有:核糖体蛋白结合位点突变、细胞膜变异、主动外排泵转运系统和修饰酶降解等。在肺炎克雷伯菌、大肠杆菌中,核糖体S10蛋白的编码基因rpsJ突变可介导替加环素耐药[47]。肠杆菌科细菌中,耐药相关外排泵AcrAB-TolC和OqxAB也是细菌对替加环素的敏感性降低的原因之一[48-49]。大肠杆菌中,由IncFIA 质粒编码的RND 型外排泵TMexCD1-TOprJ1可导致替加环素外排增加;另外,核糖体保护蛋白Tet(X)变异体结合可结合性质粒,可以在不同种属的细菌间进行替加环素耐药性的水平传播[50]。
-
头孢他啶-阿维巴坦是一种由第三代头孢菌素头孢他啶与新型β内酰胺酶抑制剂阿维巴坦组成的复合制剂,主要通过阿维巴坦抑制多种类型的β内酰胺酶保护头孢他啶,从而增强头孢他啶杀菌作用[51]。阿维巴坦对于各类β内酰胺酶有广泛的抑制活性,但对于缺乏活性位点丝氨酸残基的B类金属酶(NDM1)无抑制能力[52]。头孢他啶-阿维巴坦对于革兰阴性菌特别是肠杆菌属有较好的抗菌活性,但近年来头孢他啶-阿维巴坦耐药率逐渐上升,其耐药性产生的原因主要是产生金属β内酰胺酶:B类金属酶通过锌离子与β内酰胺类底物结合,可水解所有临床使用的丝氨酸β内酰胺酶抑制剂,包括阿维巴坦;其他原因还包括KPC型碳青霉烯酶基因过表达及β内酰胺酶关键位点氨基酸突变,还存在膜通透性缺陷(即OmpK35、OmpK36和OmpK37的改变)和青霉素结合蛋白突变、外排泵的过表达等原因。
-
美罗培南-法硼巴坦于2017年获得美国食品药品管理局(FDA)批准,2018年获得欧洲药品管理局(EMA)批准,用于治疗急性胰腺炎、复杂性腹腔感染、医院获得性肺炎、呼吸机相关性肺炎及复杂性尿路感染。法硼巴坦(RPX7009)是一种环状硼酸药效团的β-内酰胺酶抑制剂,其结构与既往上市的其他β-内酰胺酶抑制剂有所不同,法硼巴坦中的硼原子与特定β-内酰胺酶的2-巯基乙酰取代基处的催化丝氨酸形成可逆共价键而发挥作用[53]。法硼巴坦对不同碳青霉烯酶的活性不同,对Ambler A类酶(包括ESBLs、KPCs)有活性,但对B类酶(NDM、VIM、IMP)和D类酶(OXA-48)没有活性[54]。一项2018年的研究结果显示,美罗培南-法硼巴坦对共991珠的KPC阳性肠杆菌科分离物的体外敏感性为99.0%,高于头孢他啶-阿维巴坦(98.2%)和替加环素(95.8%);根据MIC90s数据可得美罗培南-法硼巴坦抗菌效力分别是头孢他啶-阿维巴坦的4倍和美罗培南的64倍[55]。一项多中心、回顾性研究指出比较头孢他啶-阿维巴坦与美罗培南-法硼巴坦治疗成人CRE感染,两种治疗方案30 d和90 d死亡率无差异,但头孢他啶-阿维巴坦单药耐药更为常见[56]。美罗培南-法硼巴坦耐药主要是由于OmpK35/36失活(孔蛋白突变)、blaKPC基因拷贝数增加、抗菌药物的靶点修饰及外排泵的激活[57-59]。
-
亚胺培南-瑞来巴坦于2019年获得FDA批准,2020年获得EMA批准,用于治疗由多重耐药革兰氏阴性菌引起的复杂性尿路感染、复杂性腹腔感染、医院获得性肺炎和呼吸机相关性肺炎。瑞来巴坦(MK-
7655 )是一种二氮杂双环辛烷抑制剂,结构上相比阿维巴坦在2位羰基上添加了一个哌啶环,其高活性来源于高应变双环脲核与吸电子氨基氧基硫酸盐,但这样的高反应导致其稳定性有限,pH值为4~8时能达到稳定;正常生理pH下,哌啶侧链可以防止瑞来巴坦的细胞外排[56],对A类(如KPC、TEM、SHV和CTX-M)碳青霉烯酶具有有效的体外活性,对B类(MBL、NDM、VIM和IMP)无活性,对D类(OXA-48)碳青霉烯酶的活性有限[54]。与其他新型β-内酰胺不同,亚胺培南-瑞来巴坦对氨苄西林敏感肠球菌和革兰氏阴性厌氧菌具有可靠的抗菌活性,使其成为多重感染患者的潜在首选药物,包括肠球菌、CR-NME和/或DTR铜绿假单胞菌[60]。在所有耐药机制中,B类和D类碳青霉烯酶的产生是CRE中亚胺培南-瑞来巴坦耐药的主要原因。另外亚胺培南-瑞来巴坦耐药也可能由其他机制共同导致,包括碳青霉烯酶突变、碳青霉烯酶过表达、青霉素结合蛋白突变或低表达、外排增加和通透性降低等[54, 57]。 -
普拉佐米星(原ACHN-490)是一种新型的半合成氨基糖苷类抗菌药物,于2018年获得美国FDA批准,用于复杂性尿路感染,尚未获得EMA的批准。普拉佐米星对肠杆菌具有广谱活性,包括产ESBL酶或多种CRE肠杆菌,包括A类(KPC),B类(VIM, IMP)和D类(OXA-48)β-内酰胺酶[60];但对于产NDM-1酶的菌株,因为它们通常产生16S rRNA甲基转移酶,所以抗菌活性变化差异较大,因此对NDM-1流行地区的临床应用可能有限[60]。氨基糖苷类抗菌药物的耐药性通常是通过氨基糖苷修饰酶(AMEs)发生的,该酶降低了药物对核糖体靶标的结合亲和力。普拉佐米星具有几种结构修饰,可阻止大多数AMEs的活性,从而降低AMEs介导的耐药风险。但普拉佐米星不能克服16s核糖体甲基转移酶引起的修饰,这些基因可以通过质粒水平转移造成普拉佐米星的耐药[61]。
-
依拉环素(原TP-434)是一种新型四环素,于2018年获得美国FDA批准,用于复杂性尿路感染,其结构类似于替加环素,通过结合核糖体30s亚基抑制细菌蛋白质合成,对除假单胞菌之外的好氧和厌氧的革兰氏阳性与阴性菌有广谱活性[62]。依拉环素对含A类(KPC)、B类(VIM,NDM-1)和D类(OXA-48)β-内酰胺酶的CRE仍有活性[63]。四环素类抗菌药物的耐药性是通过获得编码在质粒和结合转座子或整合子上的抗性基因而发生的,因此可以在物种和属之间转移。目前已知的四环素类耐药机制有4种:外排、核糖体保护蛋白、核糖体突变和酶失活。外排是导致新型四环素耐药的主要原因。依拉环素可以避开易导致其他新型四环素在肠杆菌科和不动杆菌属中耐药的TetA外排泵;虽然葡萄球菌表达的TetK泵对依拉环素有作用,但通常不引起耐药[64-65]。另外,革兰氏阴性细菌与拟杆菌属中发现的Tet(X)酶是一种对依拉环素有活性的四环素破坏酶,可在人类肠道菌群中以无症状携带的形式存在,表明依拉环素的耐药性具有可传播性[66]。
-
头孢地尔是一种新型的铁载体头孢菌素,2019年获得美国FDA批准,用于治疗复杂性尿路感染和医院获得性肺炎和呼吸机相关性肺炎。头孢地尔通过铁转运系统主动转运,以及少量膜孔蛋白通道途径逃避细菌防御系统进入细菌细胞[67]。头孢地尔进入细胞后靶向结合青霉素结合蛋白,主要是PBP-3[68],其结构C3和C7侧链的修饰使其对多数丝氨酸和金属β -内酰胺酶(包括KPC、NDM、VIM、IMP和OXA-48)具有高度稳定性,抑制肽聚糖合成并最终导致细胞死亡[69]。近年体外研究发现,头孢地尔对CRE敏感性高[70-71]。2022年一篇有关肠杆菌中头孢地尔敏感性降低机制的综述提到某些β-内酰胺酶(尤其是NDM或KPC变体)的产生,铁载体摄取系统的修饰,以及PBP靶点的罕见突变可能会成为头孢地尔耐药的原因[72]。
-
耐碳青霉烯类肠杆菌的传播是一个紧迫的公共卫生问题,对全世界的抗菌药物疗效构成威胁。氨曲南、磷霉素、多粘菌素、替加环素与头孢哌酮阿维巴坦是国内常见的治疗药物。但上述药物耐药性逐年攀升。过去几年FDA批准上市的新型β-内酰胺和β-内酰胺酶抑制剂组合,以及新型的氨基糖苷类、四环素类和头孢菌素类药物等新药的耐药率低,或可缓解耐碳青霉烯类肠杆菌的耐药困境。
Research progress on resistance mechanisms of carbapenem-resistant Enterobacteriaceae
-
摘要: 耐碳青霉烯类肠杆菌(CRE)是全球范围内日益严重的人类健康威胁。CRE通常携带多种耐药基因,限制了治疗药物选择,需要更长的治疗时间,治疗费用更高且治疗风险更大。该综述概述了CRE的流行病学、耐药机制、耐药现状以及最新治疗药物的介绍。为临床合理使用抗菌药物,耐药菌感染的防控和治疗提供依据。Abstract: Carbapenem-resistant Enterobacteriaceae (CRE) is an increasingly serious threat to human health worldwide. CRE usually carries multiple drug resistance genes, which limit the selection of therapeutic drugs, prolong treatment time, require higher treatment costs and greater treatment risks. The epidemiology, resistance mechanisms, current resistance status of CRE and the latest therapeutic drugs for CRE were summarized in this papoer, which provide a basis for the rational use of antibiotics in clinical prevention and treatment of drug-resistant bacterial infections.
-
三阴性乳腺癌(triple-negative breast cancer,TNBC)临床特点表现为转移能力强、复发率高和患者预后差,是目前威胁女性健康常见的恶性肿瘤之一[1-3]。由于其细胞表面不表达孕酮受体、雌激素受体以及表皮生长因子受体,使得常规靶向疗法对TNBC收效甚微,目前临床治疗手段仍以传统的化疗为主。然而TNBC除了不表达多种激素受体外,往往也伴随着乳腺癌基因(breast cancer gene,BRCA)等多种基因的突变[4-6],导致其成为一种高度异质性的肿瘤类型,易对化疗药物产生抗性,进一步增加了治疗难度。
代谢旺盛的肿瘤细胞能量供应高度依赖有氧糖酵解产生的ATP,越来越多的研究表明,线粒体氧化磷酸化(oxidative phosphorylation,OXPHOS)对肿瘤细胞糖类、脂类及蛋白质类三大营养物质的相互转化和氧化还原反应的平衡有着重要的作用[7-8]。在多种流行病学、临床和实验室研究中证实,糖代谢抑制剂、线粒体呼吸链阻滞剂等具有显著的抗肿瘤作用[9]。因此,在正常细胞可承受范围内,靶向破坏肿瘤细胞的糖代谢以及干扰呼吸链的电子传递,是目前肿瘤治疗的新策略。
二甲双胍以其良好的安全性和耐受性,在糖尿病临床治疗中被广泛应用[10]。越来越多的研究表明,以二甲双胍为代表的双胍类药物对多种肿瘤具有抑制作用[11-12]。苯乙双胍是比二甲双胍作用强50倍的线粒体复合物I抑制剂[13]。然而,苯乙双胍作为一种抗肿瘤药物却难以获得各国药品管理部门的批准,主要原因是其副作用会产生大量的乳酸,易引起严重的乳酸性酸血症[14-16]。因此,联用其他辅助药物以降低苯乙双胍的使用剂量,在可控的不良反应内达到有效的抗肿瘤作用,是目前肿瘤临床治疗研究的新策略。
笔者将以肿瘤细胞能量代谢为突破点,研究低剂量的苯乙双胍联合己糖激酶抑制剂2-DG对TNBC的治疗作用,为将来针对TNBC的耐药和复发而进行的临床治疗提供新的策略。
1. 材料
1.1 实验动物及细胞
40只SPF级雌性6周龄BALB/c小鼠,体质量(20±2)g,购自浙江省实验动物中心,许可证号:SCXK(浙)2016-0002。小鼠三阴性乳腺癌细胞系4T1和人三阴性乳腺癌细胞系MBA-MD-231(中国医学科学院基础医学研究所细胞资源中心)。
1.2 实验试剂
苯乙双胍(Selleck公司);2-脱氧葡萄糖(Sigma公司);RNA反转录试剂盒(ABI公司);FITC-annexin Ⅴ/PI凋亡染色试剂盒(BD公司);葡萄糖含量检测试剂盒、乳酸含量(LA)检测试剂盒(北京索莱宝科技有限公司);海马细胞线粒体压力检测试剂盒(Agilent公司)。
2. 方法
2.1 细胞分组及药物处理
分别将1×105个4T1或MDA-MB-231细胞接种到6孔板中,每组设3个复孔。实验分为空白对照组、苯乙双胍(100 μmol/L)组、2-DG(2 mmol/L)组和联用组(苯乙双胍:10 μmol/L;2-DG:200 μmol/L)。作用48 h后,用0.25%的胰蛋白酶消化细胞,获得单细胞悬液,用于后续的细胞增殖和凋亡检测。
2.2 流式细胞仪检测细胞凋亡
药物处理细胞48 h后,收集各组细胞悬液,4℃ 500×g离心5 min。弃上清液,每管加入1 ml冷PBS重悬细胞,离心后弃上清液,清洗细胞。加入100 μl 1×偶联缓冲液重悬细胞,然后每管分别加入2 μl annexin Ⅴ和PI,4℃避光孵育30 min。加入400 μl PBS重悬细胞,流式细胞仪检测各组细胞凋亡的情况。
2.3 葡萄糖和乳酸浓度的测定
各组细胞处理48 h后,收集细胞培养上清液,分别用葡萄糖含量检测试剂盒和乳酸含量检测试剂盒测定葡萄糖和乳酸浓度,同时收集细胞并计数。
2.4 线粒体耗氧量(OCR)的测定
用海马细胞线粒体压力检测试剂盒测定4T1或MDA-MB-231细胞的线粒体OCR。细胞用药物预处理24 h,在评估前8 h,以6×105个细胞/孔的最佳培养密度将细胞转移到XF微板上,使细胞贴壁。用调节好pH 7.4的培养基对细胞进行清洗,在无CO2培养箱中平衡1 h。ABC孔分别加入寡霉素、线粒体解偶联剂(FCCP)、鱼藤酮和抗霉素A后,上机检测。
2.5 动物实验的分组及处理
分为空白(PBS)组、苯乙双胍(1 mg/kg)组、2-DG(5 mg/kg)组和联用组(苯乙双胍:0.1 mg/kg;2-DG:0.5 mg/kg),每组10只小鼠。1×105个 4T1细胞原位接种到BALB/c小鼠乳腺脂肪垫内,待肿瘤生长至5 mm×5 mm左右时,荷瘤小鼠开始给药治疗,并每天测量肿瘤的大小。各给药组小鼠按50 μl体积瘤内注射给予,对照组给予等体积的PBS,每2 d给药1次。连续给药10次后,观察荷瘤小鼠的肿瘤大小并记录各组小鼠的死亡时间。
3. 统计学分析
采用SPSS 22.0软件进行统计学分析。实验数据用(
$\bar x $ ±s )表示,两组间比较用t检验,多组的组间比较采用单因素方差分析,动物生存时间比较采用Kaplan-Meier法。4. 结果
4.1 苯乙双胍下调4T1或MDA-MB-231细胞线粒体OCR
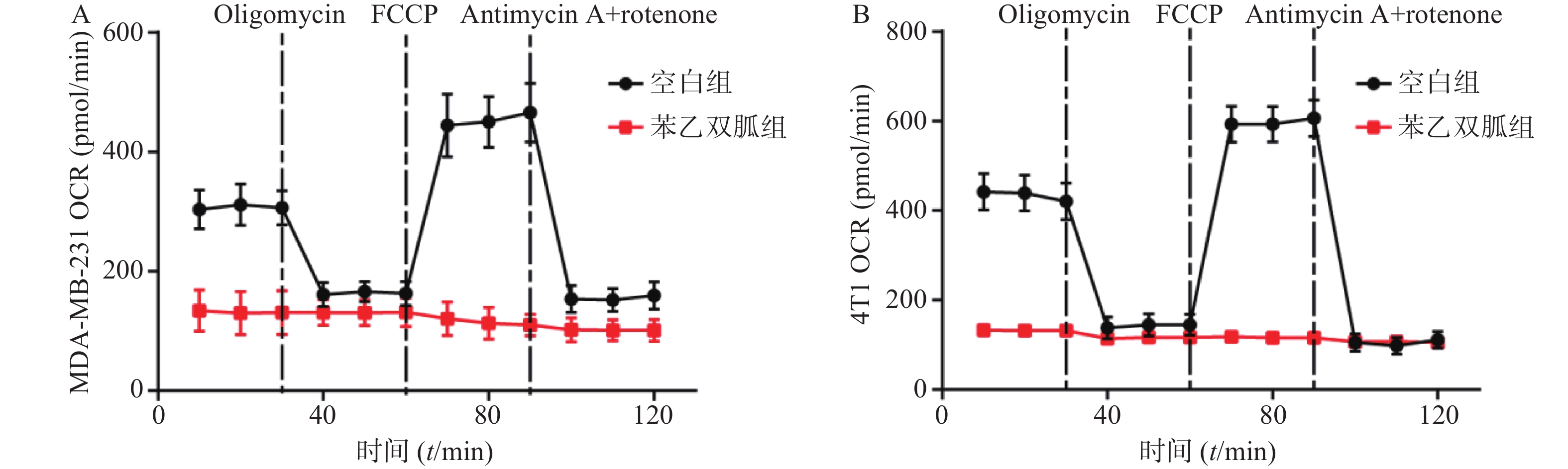
为检测苯乙双胍对线粒体呼吸的影响,将100 μmol/L苯乙双胍分别作用于4T1和MDA-MB-231细胞,24 h后收获细胞,海马生物能量分析仪检测细胞OCR水平。结果表明,苯乙双胍孵育24 h后,MDA-MB-231或4T1细胞线粒体OCR与空白组相比显著降低(图1)。
4.2 苯乙双胍对4T1或MDA-MB-231细胞有氧糖酵解的影响
100 μmol/L苯乙双胍处理4T1和MDA-MB-231细胞48 h后,检测细胞上清液中己糖激酶表达量、葡萄糖以及乳酸浓度。结果显示,4T1和MDA-MB-231细胞上清液中己糖激酶表达量,苯乙双胍给药组(4.6±0.17,3.73±0.21,n=3)明显高于空白组(1±0.15,1±0.12, n=3),组间有显著性差异(P<0. 001),表明苯乙双胍可在基因水平显著上调己糖激酶的表达(图2A);4T1和MDA-MB-231细胞上清中葡萄糖消耗量,苯乙双胍给药组(356±31,397±42,n=3)μg/105个细胞明显高于空白组(289±25,301±32,n=3)μg/105细胞,组间有显著性差异(P < 0. 05),表明苯乙双胍能促进细胞摄取更多的葡萄糖,致使培养基中葡萄糖含量显著增加(图2B);4T1和MDA-MB-231细胞上清中乳酸浓度,苯乙双胍给药组(5.59±0.52, 7.83±0.78, n=3)μmol/L明显高于空白组(2.37±0.18,4.01±0.45,n=3)μmol/L,组间有显著性差异(P < 0.01),表明苯乙双胍能显著促进培养上清液中乳酸的产生(图2C)。
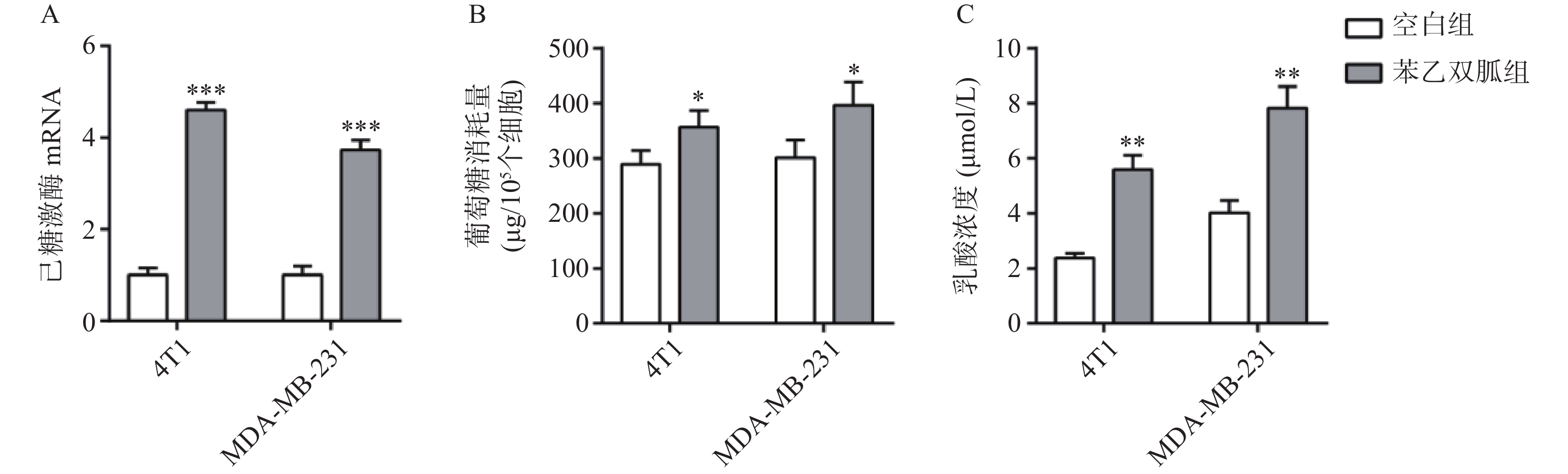 图 2 苯乙双胍对MDA-MB-231和4T1细胞有氧糖酵解的影响 (`x±s , n = 3)注:A. 己糖激酶mRNA表达量;B. 葡萄糖消耗量;C. 乳酸浓度;*P<0.05, ** P<0.01,*** P < 0. 001 ,与空白组比较
图 2 苯乙双胍对MDA-MB-231和4T1细胞有氧糖酵解的影响 (`x±s , n = 3)注:A. 己糖激酶mRNA表达量;B. 葡萄糖消耗量;C. 乳酸浓度;*P<0.05, ** P<0.01,*** P < 0. 001 ,与空白组比较4.3 苯乙双胍联合2-DG诱导三阴性乳腺癌细胞凋亡
由于苯乙双胍可上调4T1和MDA-MB-231细胞的有氧糖酵解,因此当我们联用低剂量的己糖激酶抑制剂2-DG时,三阴性乳腺癌细胞会发生什么变化呢?结果显示,与空白组相比,单用苯乙双胍或2-DG均能显著降低4T1与MDA-MB-231细胞存活率(P<0.01)(表1,图3);与苯乙双胍或2-DG单药组相比,苯乙双胍联用2-DG,即使降低90%剂量,仍然可以显著降低4T1与MDA-MB-231细胞的存活率(P<0.001)(表1,图3),以上结果表明,苯乙双胍联用2-DG能显著促进三阴性乳腺癌细胞的凋亡。与此同时,检测细胞上清液中的乳酸含量,相比苯乙双胍组(5.59±0.52,7.83±0.78,n=3)μmol/L,苯乙双胍与2-DG联用组(3.46±0.37,5.18±0.62,n=3)μmol/L细胞的乳酸产量也大幅下降(P < 0.01)(图4)。
表 1 苯乙双胍联合2-DG对三阴性乳腺癌细胞凋亡的影响[`x±s , n = 3,存活率(%)]组别 4T1 MDA-MB-231 空白组(PBS) 96.37±2.31 97.63±1.46 苯乙双胍组(100 μmol/L) 86.70±1.83 * 85.53±1.46 ** 2-DG(2 mmol/L) 81.27±2.16** 80.67±4.07** 苯乙双胍+2-DG组(苯乙双胍:
10 μmol/L,2-DG: 200 μmol/L)64.63±2.28*** 51.97±2.29*** *P<0. 05,**P<0.01,***P<0.001,与空白组比较 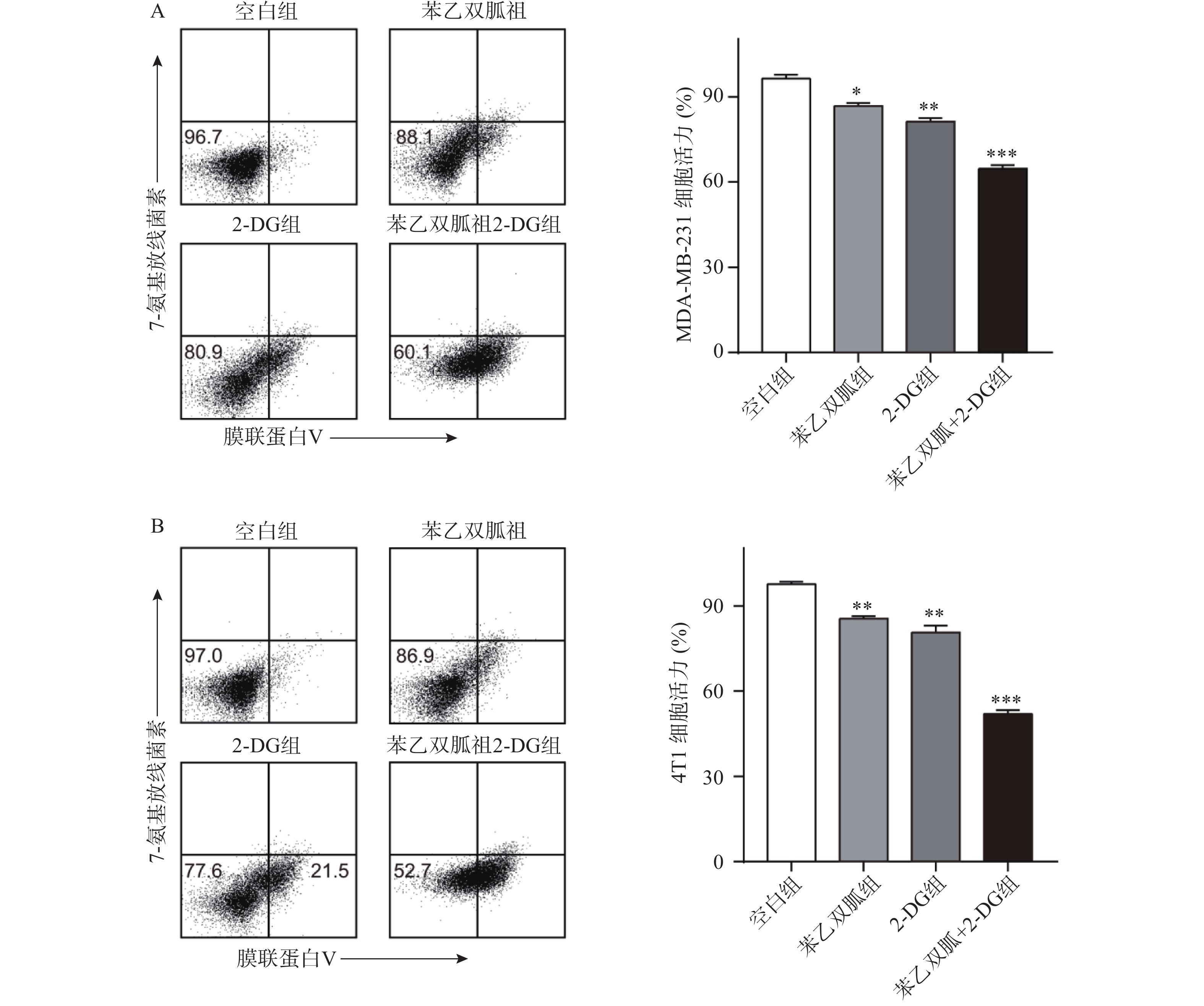 图 3 苯乙双胍联合2-DG对三阴性乳腺癌细胞凋亡的影响 (`x±s , n=3)注:A. 苯乙双胍(100 μmol/L)、2-DG(2 mmol/L)、苯乙双胍(100 ng/ml)联合2-DG(20 nmol/L)处理MDA-MB-231,流式细胞仪检测细胞凋亡情况;B. 苯乙双胍(100 μmol/L)、2-DG(2 mmol/L)、苯乙双胍(100 ng/ml)联合2-DG(20 nmol/L)处理4T1,流式细胞仪检测细胞凋亡情况;* P < 0. 05, ** P<0. 01, *** P<0. 001,与空白组比较
图 3 苯乙双胍联合2-DG对三阴性乳腺癌细胞凋亡的影响 (`x±s , n=3)注:A. 苯乙双胍(100 μmol/L)、2-DG(2 mmol/L)、苯乙双胍(100 ng/ml)联合2-DG(20 nmol/L)处理MDA-MB-231,流式细胞仪检测细胞凋亡情况;B. 苯乙双胍(100 μmol/L)、2-DG(2 mmol/L)、苯乙双胍(100 ng/ml)联合2-DG(20 nmol/L)处理4T1,流式细胞仪检测细胞凋亡情况;* P < 0. 05, ** P<0. 01, *** P<0. 001,与空白组比较4.4 苯乙双胍联用2-DG显著抑制4T1肿瘤的生长
为了进一步验证苯乙双胍联合2-DG在体内抗肿瘤的效果,我们将4T1细胞原位接种到小鼠乳腺脂肪垫中,并分别给予单药治疗或联合治疗,观察肿瘤的生长速度以及荷瘤小鼠的生存时间。结果显示,与苯乙双胍或2-DG单药组相比,苯乙双胍联合2-DG组可显著抑制荷瘤小鼠体内肿瘤的生长速度(P<0.01)(图5A)。此外,苯乙双胍联合2-DG组荷瘤小鼠中位生存时间为72.5 d,高于苯乙双胍组(57 d)、2-DG组(55.5 d)、空白组组(50.5 d),差异有统计学意义(P<0.01)(图5B),表明苯乙双胍联合2-DG可以延长荷瘤小鼠生存时间。
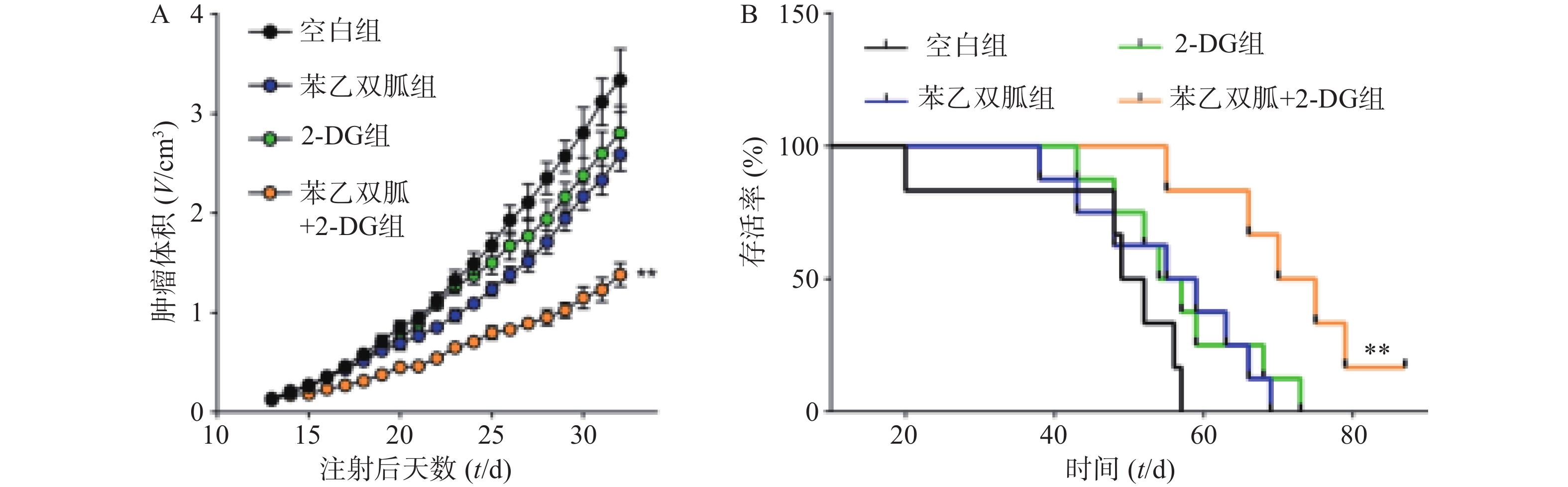 图 5 苯乙双胍联合2-DG对4T1细胞造模小鼠肿瘤体积和生存时间的影响(`x±s , n=10)注:A. 1×105个 4T1细胞原位接种到BALB/c小鼠乳腺脂肪垫内,待肿瘤生长至5 mm×5 mm左右时,荷瘤小鼠开始给药治疗,测量每日肿瘤体积,**P <0.01,与苯乙双胍组比较;B. 4T1细胞造模小鼠给药后的生存时间;**P<0.01,与2-DG组比较
图 5 苯乙双胍联合2-DG对4T1细胞造模小鼠肿瘤体积和生存时间的影响(`x±s , n=10)注:A. 1×105个 4T1细胞原位接种到BALB/c小鼠乳腺脂肪垫内,待肿瘤生长至5 mm×5 mm左右时,荷瘤小鼠开始给药治疗,测量每日肿瘤体积,**P <0.01,与苯乙双胍组比较;B. 4T1细胞造模小鼠给药后的生存时间;**P<0.01,与2-DG组比较5. 讨论
二甲双胍等双胍类糖尿病治疗药物,能够通过抑制线粒体复合物I来降低细胞内ATP水平。线粒体复合物I的损伤会降低NADH氧化为NAD+,这是维持TCA循环功能的关键反应,并最终导致抑制氧化磷酸化。前期研究表明,二甲双胍表现出显著的抗肿瘤作用。苯乙双胍与二甲双胍具有非常相似的代谢特征,而苯乙双胍的效力更强[17-18]。由于乳酸性酸中毒病死率高,2型糖尿病临床治疗不再使用苯乙双胍作为一线药物[19]。 然而作为一种抗癌药物,因其较低的有效剂量和较短的疗程,与糖尿病临床治疗大有不同,苯乙双胍被认为最有希望替代二甲双胍的双胍类药物[20-22]。
本研究采用小鼠三阴性乳腺癌细胞系4T1和人三阴性乳腺癌细胞系MBA-MD-231作为研究对象,发现苯乙双胍可显著抑制其线粒体氧化磷酸化,并上调肿瘤细胞的糖酵解。当加入己糖激酶抑制剂2-DG时,糖酵解途径被阻断,肿瘤细胞被迫使用氧化磷酸化来获取ATP,在此情况下,细胞对苯乙双胍更加敏感。基于该项发现,采用苯乙双胍联用2-DG治疗三阴性乳腺癌细胞。动物体内外结果表明,苯乙双胍联用2-DG可显著增加4T1细胞和MBA-MD-231细胞的死亡率,并延长荷瘤小鼠的生存时间。除了对这两种糖代谢方式双重抑制作用外,联用2-DG带来的另一个优势是可大大降低苯乙双胍的使用剂量,从而减轻苯乙双胍代谢产生的乳酸对机体的不良作用。
综上所述,通过苯乙双胍联用2-DG,可显著增强苯乙双胍对三阴性乳腺癌的凋亡作用,并降低其使用剂量,减轻不良反应,这一发现为三阴性乳腺癌的临床治疗提供新的策略。
-
[1] World Health Organization. Global antimicrobial resistance and use surveillance system (GLASS) report:2022 [R]. http:// www.who.int/news-room/fact-sheets/detail/antibiotic-resistance. [2] PAPP-WALLACE K M, ENDIMIANI A, TARACILA M A, et al. Carbapenems: past, present, and future[J]. Antimicrob Agents Chemother, 2011, 55(11):4943-4960. doi: 10.1128/AAC.00296-11 [3] DING L, SHEN S Q, CHEN J, et al. Klebsiella pneumoniae carbapenemase variants: the new threat to global public health[J]. Clin Microbiol Rev, 2023, 36(4):e0000823. doi: 10.1128/cmr.00008-23 [4] Centers for Disease Control and Prevention. Antibiotic resistance threats in the United States, 2019 (2019 AR Threats Report) [R]. https//www.cdc.gov/antimicrobial-resistance/data-research/threats/?CDC_AAref_Val=https://www.cdc.gov/drugresistance/biggest-threats.html. [5] GUH A Y, LIMBAGO B M, KALLEN A J. Epidemiology and prevention of carbapenem-resistant Enterobacteriaceae in the United States[J]. Expert Rev Anti Infect Ther, 2014, 12(5):565-580. doi: 10.1586/14787210.2014.902306 [6] SADER H S, CASTANHEIRA M, FLAMM R K, et al. Tigecycline activity tested against carbapenem-resistant Enterobacteriaceae from 18 European nations: results from the SENTRY surveillance program (2010–2013)[J]. Diagn Microbiol Infect Dis, 2015, 83(2):183-186. doi: 10.1016/j.diagmicrobio.2015.06.011 [7] TOMPKINS K, VAN DUIN D. Treatment for carbapenem-resistant Enterobacterales infections: recent advances and future directions[J]. Eur J Clin Microbiol Infect Dis, 2021, 40(10):2053-2068. doi: 10.1007/s10096-021-04296-1 [8] PALZKILL T. Structural and mechanistic basis for extended-spectrum drug-resistance mutations in altering the specificity of TEM, CTX-M, and KPC β-lactamases[J]. Front Mol Biosci, 2018, 5(5):16. doi: 10.3389/fmolb.2018.00016 [9] WALTHER-RASMUSSEN J, HØIBY N. Class A carbapenemases[J]. J Antimicrob Chemother, 2007, 60(3):470-482. doi: 10.1093/jac/dkm226 [10] HOSSAIN A, FERRARO M J, PINO R M, et al. Plasmid-mediated carbapenem-hydrolyzing enzyme KPC-2 in an Enterobacter sp[J]. Antimicrob Agents Chemother, 2004, 48(11):4438-4440. doi: 10.1128/AAC.48.11.4438-4440.2004 [11] TOOKE C L, HINCHLIFFE P, BRAGGINTON E C, et al. β-lactamases and β-lactamase inhibitors in the 21st century[J]. J Mol Biol, 2019, 431(18):3472-3500. doi: 10.1016/j.jmb.2019.04.002 [12] POTTER R F, D’SOUZA A W, DANTAS G. The rapid spread of carbapenem-resistant Enterobacteriaceae[J]. Drug Resist Updat, 2016, 29:30-46. doi: 10.1016/j.drup.2016.09.002 [13] WALSH T R, TOLEMAN M A, POIREL L, et al. Metallo-beta-lactamases: the quiet before the storm?[J]. Clin Microbiol Rev, 2005, 18(2):306-325. doi: 10.1128/CMR.18.2.306-325.2005 [14] MAIRI A, PANTEL A, SOTTO A, et al. OXA-48-like carba-penemases producing Enterobacteriaceae in different niches[J]. Eur J Clin Microbiol Infect Dis, 2018, 37(4):587-604. doi: 10.1007/s10096-017-3112-7 [15] PITOUT J D D, PEIRANO G, KOCK M M, et al. The global ascendency of OXA-48-type carbapenemases[J]. Clin Microbiol Rev, 2019, 33(1):e00102-e00119. [16] GUZMÁN-PUCHE J, JENAYEH R, PÉREZ-VÁZQUEZ M, et al. Characterization of OXA-48-producing Klebsiella oxytoca isolates from a hospital outbreak in Tunisia[J]. J Glob Antimicrob Resist, 2021, 24:306-310. doi: 10.1016/j.jgar.2021.01.008 [17] HEIREMAN L, HAMERLINCK H, VANDENDRIESSCHE S, et al. Toilet drain water as a potential source of hospital room-to-room transmission of carbapenemase-producing Klebsiella pneumoniae[J]. J Hosp Infect, 2020, 106(2):232-239. doi: 10.1016/j.jhin.2020.07.017 [18] SHAIDULLINA E, SHELENKOV A, YANUSHEVICH Y, et al. Antimicrobial resistance and genomic characterization of OXA-48- and CTX-M-15-co-producing hypervirulent Klebsiella pneumoniae ST23 recovered from nosocomial outbreak[J]. Antibiotics(Basel), 2020, 9(12):862. [19] ZHANG R, LIU L Z, ZHOU H W, et al. Nationwide surveillance of clinical carbapenem-resistant Enterobacteriaceae (CRE) strains in China[J]. EBioMedicine, 2017, 19(5):98-106. doi: 10.1016/j.ebiom.2017.04.032 [20] WANG Q, WANG X J, WANG J, et al. Phenotypic and genotypic characterization of Carbapenem-resistant Enterobacteriaceae: data from a longitudinal large-scale cre study in China (2012−2016)[J]. Clin Infect Dis, 2018, 67(suppl_2):S196-S205. doi: 10.1093/cid/ciy660 [21] HAN R R, SHI Q Y, WU S, et al. Dissemination of carbapenemases(KPC, NDM, OXA-48, IMP, and VIM)among carbapenem-resistant Enterobacteriaceae isolated from adult and children patients in China[J]. Front Cell Infect Microbiol, 2020, 10:314. doi: 10.3389/fcimb.2020.00314 [22] 莫银竹, 宋沧桑, 李志伟, 等. 耐碳青霉烯肺炎克雷伯菌耐药机制及治疗策略的研究进展[J]. 中国药物评价, 2023, 40(3):217-223. [23] 孟文凯, 包志瑶, 李庆云. 肠杆菌科细菌AcrAB-TolC外排泵调控机制及对策的研究进展[J]. 中国药物与临床, 2021, 21(19):3280-3282. [24] WESTON N, SHARMA P, RICCI V, et al. Regulation of the AcrAB-TolC efflux pump in Enterobacteriaceae[J]. Res Microbiol, 2018, 169(7-8):425-431. doi: 10.1016/j.resmic.2017.10.005 [25] OLLIVER A, VALLÉ M, CHASLUS-DANCLA E, et al. Role of an acrR mutation in multidrug resistance of in vitro-selected fluoroquinolone-resistant mutants of Salmonella enterica serovar Typhimurium[J]. FEMS Microbiol Lett, 2004, 238(1):267-272. [26] WEBBER M A, TALUKDER A, PIDDOCK L J. Contribution of mutation at amino acid 45 of AcrR to acrB expression and ciprofloxacin resistance in clinical and veterinary Escherichia coli isolates[J]. Antimicrob Agents Chemother, 2005, 49(10):4390-4392. doi: 10.1128/AAC.49.10.4390-4392.2005 [27] LI X Z, PLÉSIAT P, NIKAIDO H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria[J]. Clin Microbiol Rev, 2015, 28(2):337-418. doi: 10.1128/CMR.00117-14 [28] VALENTIN-HANSEN P, JOHANSEN J, RASMUSSEN A A. Small RNAs controlling outer membrane porins[J]. Curr Opin Microbiol, 2007, 10(2):152-155. doi: 10.1016/j.mib.2007.03.001 [29] WANG X D, CAI J C, ZHOU H W, et al. Reduced susceptibility to carbapenems in Klebsiella pneumoniae clinical isolates associated with plasmid-mediated beta-lactamase production and OmpK36 porin deficiency[J]. J Med Microbiol, 2009, 58(Pt 9): 1196-1202. [30] BORNET C, DAVIN-REGLI A, BOSI C, et al. Imipenem resistance of Enterobacter aerogenes mediated by outer membrane permeability[J]. J Clin Microbiol, 2000, 38(3):1048-1052. doi: 10.1128/JCM.38.3.1048-1052.2000 [31] KUMAR G, GALANIS C, BATCHELDER HR, et al. Penicillin binding proteins and β-lactamases of mycobacterium tuberculosis: reexamination of the historical paradigm[J]. mSphere, 2022, 7(1): e0003922. [32] 刘洁, 赵建平. 碳青霉烯耐药革兰阴性杆菌的耐药机制及抗菌药物的研究进展[J]. 国外医药(抗生素分册), 2024, 45(1):20-27. [33] JEAN S S, LEE W S, LAM C, et al. Carbapenemase-producing Gram-negative bacteria: current epidemics, antimicrobial susceptibility and treatment options[J]. Future Microbiol, 2015, 10(3):407-425. doi: 10.2217/fmb.14.135 [34] RACT P, COMPAIN F, ROBIN F, et al. Synergistic in vitro activity between aztreonam and amoxicillin-clavulanate against Enterobacteriaceae-producing class B and/or class D carbapenemases with or without extended-spectrum β-lactamases[J]. J Med Microbiol, 2019, 68(9):1292-1298. doi: 10.1099/jmm.0.001052 [35] MARAKI S, MAVROMANOLAKI V E, MORAITIS P, et al. Ceftazidime-avibactam, meropenen-vaborbactam, and imipenem-relebactam in combination with aztreonam against multidrug-resistant, metallo-β-lactamase-producing Klebsiella pneumoniae[J]. Eur J Clin Microbiol Infect Dis, 2021, 40(8):1755-1759. doi: 10.1007/s10096-021-04197-3 [36] FALAGAS M E, VOULOUMANOU E K, SAMONIS G, et al. Fosfomycin[J]. Clin Microbiol Rev, 2016, 29(2):321-347. doi: 10.1128/CMR.00068-15 [37] ESCHENBURG S, PRIESTMAN M, SCHÖNBRUNN E. Evidence that the fosfomycin target Cys115 in UDP-N-acetylglucosamine enolpyruvyl transferase (MurA) is essential for product release[J]. J Biol Chem, 2005, 280(5):3757-3763. doi: 10.1074/jbc.M411325200 [38] DE OLIVEIRA M V D, FURTADO R M, DA COSTA K S, et al. Advances in UDP-N-acetylglucosamine enolpyruvyl transferase(MurA) covalent inhibition[J]. Front Mol Biosci, 2022, 9:889825. doi: 10.3389/fmolb.2022.889825 [39] 靳迺诗, 何菊英, 冯伟, 等. 磷霉素在多药耐药肠杆菌科细菌感染治疗中的研究进展[J]. 实用药物与临床, 2022, 25(6):568-572. [40] ITO R, MUSTAPHA M M, TOMICH A D, et al. Widespread fosfomycin resistance in gram-negative bacteria attributable to the chromosomal fosA gene[J]. mBio, 2017, 8(4):e00749-e00717. [41] HUANG L, CAO M, HU Y Y, et al. Prevalence and mechanisms of fosfomycin resistance among KPC-producing Klebsiella pneumoniae clinical isolates in China[J]. Int J Antimicrob Agents, 2021, 57(1):106226. doi: 10.1016/j.ijantimicag.2020.106226 [42] MOTSCH J, MURTA DE OLIVEIRA C, STUS V, et al. RESTORE-IMI 1: a multicenter, randomized, double-blind trial comparing efficacy and safety of Imipenem/Relebactam vs colistin plus imipenem in patients with Imipenem-nonsusceptible bacterial infections[J]. Clin Infect Dis, 2020, 70(9):1799-1808. doi: 10.1093/cid/ciz530 [43] WUNDERINK R G, GIAMARELLOS-BOURBOULIS E J, RAHAV G, et al. Effect and safety of meropenem-vaborbactam versus best-available therapy in patients with carbapenem-resistant Enterobacteriaceae infections: the TANGO II randomized clinical trial[J]. Infect Dis Ther, 2018, 7(4):439-455. doi: 10.1007/s40121-018-0214-1 [44] OLOWO-OKERE A, YACOUBA A. Molecular mechanisms of colistin resistance in Africa: a systematic review of literature[J]. Germs, 2020, 10(4):367-379. doi: 10.18683/germs.2020.1229 [45] EICHENBERGER E M, THADEN J T. Epidemiology and mechanisms of resistance of extensively drug resistant gram-negative bacteria[J]. Antibiotics(Basel), 2019, 8(2):37. [46] 周玉, 李玉茹, 邓新立, 等. 临床常见肠杆菌科细菌对替加环素耐药机制研究进展[J]. 中华医院感染学杂志, 2023, 33(2):310-315. [47] YAGHOUBI S, ZEKIY AO, KRUTOVA M, et al. Tigecycline antibacterial activity, clinical effectiveness, and mechanisms and epidemiology of resistance: narrative review[J]. Eur J Clin Microbiol Infect Dis, 2022, 41(7): 1003-1022. [48] POURNARAS S, KOUMAKI V, SPANAKIS N, et al. Current perspectives on tigecycline resistance in Enterobacteriaceae: susceptibility testing issues and mechanisms of resistance[J]. Int J Antimicrob Agents, 2016, 48(1):11-18. doi: 10.1016/j.ijantimicag.2016.04.017 [49] YOON E J, OH Y, JEONG S H. Development of tigecycline resistance in carbapenemase-producing Klebsiella pneumoniae sequence type 147 via AcrAB overproduction mediated by replacement of the ramA promoter[J]. Ann Lab Med, 2020, 40(1):15-20. doi: 10.3343/alm.2020.40.1.15 [50] LI Y, SUN X R, XIAO X, et al. Global distribution and genomic characteristics of Tet(X)-positive Escherichia coli among humans, animals, and the environment[J]. Sci Total Environ, 2023, 887:164148. doi: 10.1016/j.scitotenv.2023.164148 [51] SHIRLEY M. Ceftazidime-avibactam: a review in the treatment of serious gram-negative bacterial infections[J]. Drugs, 2018, 78(6):675-692. doi: 10.1007/s40265-018-0902-x [52] GIACOBBE D R, BASSETTI M. Innovative β-lactam/β-lactamase inhibitor combinations for carbapenem-resistant Gram-negative bacteria[J]. Future Microbiol, 2022, 17:393-396. doi: 10.2217/fmb-2021-0301 [53] LEE Y R, BAKER N T. Meropenem-vaborbactam: a carbapenem and beta-lactamase inhibitor with activity against carbapenem-resistant Enterobacteriaceae[J]. Eur J Clin Microbiol Infect Dis, 2018, 37(8):1411-1419. doi: 10.1007/s10096-018-3260-4 [54] ZHANEL G G, LAWRENCE C K, ADAM H, et al. Imipenem-relebactam and meropenem-vaborbactam: two novel carbapenem-β-lactamase inhibitor combinations[J]. Drugs, 2018, 78(1):65-98. doi: 10.1007/s40265-017-0851-9 [55] HACKEL M A, LOMOVSKAYA O, DUDLEY M N, et al. In vitro activity of meropenem-vaborbactam against clinical isolates of KPC-positive Enterobacteriaceae[J]. Antimicrob Agents Chemother, 2018, 62(1):e01904-e01917. [56] ACKLEY R, ROSHDY D, MEREDITH J, et al. Meropenem-vaborbactam versus ceftazidime-avibactam for treatment of carbapenem-resistant Enterobacteriaceae infections[J]. Antimicrob Agents Chemother, 2020, 64(5):e02313-e02319. [57] GAIBANI P, GIANI T, BOVO F, et al. Resistance to ceftazidime/avibactam, meropenem/vaborbactam and imipenem/relebactam in gram-negative MDR bacilli: molecular mechanisms and susceptibility testing[J]. Antibiotics(Basel), 2022, 11(5):628. [58] SUN D X, RUBIO-APARICIO D, NELSON K, et al. Meropenem-vaborbactam resistance selection, resistance prevention, and molecular mechanisms in mutants of KPC-producing Klebsiella pneumoniae[J]. Antimicrob Agents Chemother, 2017, 61(12):e01694-e01617. [59] MO Y, LORENZO M, FARGHALY S, et al. What’s new in the treatment of multidrug-resistant gram-negative infections?[J]. Diagn Microbiol Infect Dis, 2019, 93(2):171-181. doi: 10.1016/j.diagmicrobio.2018.08.007 [60] ELJAALY K, ALHARBI A, ALSHEHRI S, et al. Plazomicin: a novel aminoglycoside for the treatment of resistant gram-negative bacterial infections[J]. Drugs, 2019, 79(3):243-269. doi: 10.1007/s40265-019-1054-3 [61] SARAVOLATZ L D, STEIN G E. Plazomicin: a new aminoglycoside[J]. Clin Infect Dis, 2020, 70(4):704-709. [62] ZHANEL G G, CHEUNG D, ADAM H, et al. Review of eravacycline, a novel fluorocycline antibacterial agent[J]. Drugs, 2016, 76(5):567-588. doi: 10.1007/s40265-016-0545-8 [63] ZHANG Y L, LIN X Y, BUSH K. In vitro susceptibility of β-lactamase-producing carbapenem-resistant Enterobacteriaceae (CRE) to eravacycline[J]. J Antibiot, 2016, 69(8):600-604. doi: 10.1038/ja.2016.73 [64] LIVERMORE D M, MUSHTAQ S, WARNER M, et al. In vitro activity of eravacycline against carbapenem-resistant Enterobacteriaceae and Acinetobacter baumannii[J]. Antimicrob Agents Chemother, 2016, 60(6):3840-3844. doi: 10.1128/AAC.00436-16 [65] GROSSMAN T H, STAROSTA A L, FYFE C, et al. Target- and resistance-based mechanistic studies with TP-434, a novel fluorocycline antibiotic[J]. Antimicrob Agents Chemother, 2012, 56(5):2559-2564. doi: 10.1128/AAC.06187-11 [66] ALOSAIMY S, ABDUL-MUTAKABBIR J C, KEBRIAEI R, et al. Evaluation of eravacycline: a novel fluorocycline[J]. Pharmacotherapy, 2020, 40(3):221-238. doi: 10.1002/phar.2366 [67] KAYE K S, NAAS T, POGUE J M, et al. Cefiderocol, a siderophore cephalosporin, as a treatment option for infections caused by carbapenem-resistant enterobacterales[J]. Infect Dis Ther, 2023, 12(3):777-806. doi: 10.1007/s40121-023-00773-6 [68] SATO T, YAMAWAKI K. Cefiderocol: discovery, chemistry, and in vivo profiles of a novel siderophore cephalosporin[J]. Clin Infect Dis, 2019, 69(Suppl 7):S538-S543. [69] EL-LABABIDI R M, RIZK J G. Cefiderocol: a siderophore cephalosporin[J]. Ann Pharmacother, 2020, 54(12):1215-1231. doi: 10.1177/1060028020929988 [70] SHORTRIDGE D, STREIT J M, MENDES R, et al. In vitro activity of cefiderocol against U. S. and European gram-negative clinical isolates collected in 2020 as part of the SENTRY antimicrobial surveillance program[J]. Microbiol Spectr, 2022, 10(2):e0271221. doi: 10.1128/spectrum.02712-21 [71] LONGSHAW C, MANISSERO D, TSUJI M, et al. In vitro activity of the siderophore cephalosporin, cefiderocol, against molecularly characterized, carbapenem-non-susceptible Gram-negative bacteria from Europe[J]. JAC Antimicrob Resist, 2020, 2(3):dlaa060. doi: 10.1093/jacamr/dlaa060 [72] KARAKONSTANTIS S, ROUSAKI M, KRITSOTAKIS E I. Cefiderocol: systematic review of mechanisms of resistance, heteroresistance and in vivo emergence of resistance[J]. Antibiotics(Basel), 2022, 11(6):723. 期刊类型引用(3)
1. 杨凌,韩杰,谢小中,金万清,彭清林. 基于巨噬细胞及其相关通路探讨骨坚散干预激素性股骨头坏死作用机制. 辽宁中医药大学学报. 2025(02): 113-118 .  百度学术
百度学术2. 刘奇,章越,盛燕,胡云莉,姜慧洁,慎凯峰,姜艳,陈承守,周丹英. 基于层次分析法-熵权法-独立性权法结合正交设计法优选胃病1号提取工艺. 中国现代应用药学. 2023(21): 2998-3004 . 百度学术3. 陆玫霖,潘其明,王宝林,何宇铭,黄婉凤,陈明,钟国跃,杨世林,高红伟. 山鸡椒的化学成分、药理活性及临床应用研究进展. 中草药. 2022(17): 5565-5581 . 百度学术其他类型引用(2)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 6286
- HTML全文浏览量: 2324
- PDF下载量: 71
- 被引次数: 5
 下载:
下载:
