

 下载:
下载:

 下载:
下载:

-
雷公藤为卫矛科雷公藤属植物雷公藤Tripterygium wilfordii Hook.f.的干燥根,是一种公认的同时具有较强药效和较强毒性的中药材,广泛用于治疗类风湿性关节炎、肾病综合征、系统性红斑狼疮等疾病[1]。但其毒性较大,不良反应高发且严重,常使患者不能耐受[2]。雷公藤的主要有效成分为二萜类、三萜类和生物碱类,研究表明各种成分均具有不同程度的抗炎、抗肿瘤和免疫抑制等活性[3-4],其中生物碱的毒性要小于二萜和三萜类成分,临床应用具有疗效好,不良反应较小的特点[5]。雷公藤次碱是雷公藤生物碱中含量较高的一种倍半萜类单体化合物。目前研究显示其具有良好的杀虫活性,其他药理活性和机理研究较少,特别是抗炎和免疫抑制活性方面。
本研究主要通过建立脂多糖(LPS)诱导的RAW264.7细胞炎症模型,探讨雷公藤次碱对LPS诱导的炎性因子NO,IL-1β,TNF-α和IL-6释放的影响,用免疫印迹法考察雷公藤次碱对TRAF6、IRAK、NF-κB、IκBα、JNK、ERK、p38等关键蛋白表达或磷酸化的影响,探讨其可能的抗炎作用机制,为促进雷公藤次碱的应用研究提供基础。
-
雷公藤次碱(纯度98%,上海纯优生物科技有限公司);DMEM细胞培养基、胎牛血清(Life Technologies GIBCO);Griess试剂、细胞计数盒-8 (CCK-8)(上海碧云天生物技术有限公司);脂多糖(LPS, Sigma-Aldrich);IL-1β、TNF-α和IL-6检测试剂盒(达科为生物技术有限公司);核质提取试剂盒(Dojindo Laboratories);TRAF6、IRAF、p-IRAF、p-p38、p38、p-JNK、JNK、p-ERK、ERK、p-IκBα、IκBα、NF-κB p65、β-actin、HRP山羊抗兔抗体(Cell Signaling Technology);RAW264.7细胞购自中国科学院上海细胞库。
-
RAW264.7细胞在含10%的新生小牛血清及100 U/ml青霉素、链霉素的DMEM高糖培养液中进行培养,培养条件为5%CO2、37 ℃,隔天换液,每日观察细胞的生长状况。实验时取对数生长期RAW264.7细胞,胰酶消化,加入新鲜培养基反复吹打成细胞混悬液,细胞增殖实验、细胞因子分泌含量测定调整细胞浓度为1×105 cells/ml,以每孔100 μl细胞悬液接种于96孔细胞培养板中,细胞蛋白表达测定调整细胞浓度到2.25×105 cells/ml,以每孔2 ml接种于6孔细胞培养板中,然后细胞置于37 ℃、5%CO2培养箱中培养过夜。在药物处理前2 h给予LPS溶液(使其终浓度为1 μg/ml)诱导RAW264.7细胞发生炎症反应建立模型。设空白组(不给予LPS和药物处理)、模型组(给予LPS处理)、药物组(给予LPS诱导后再给予相应浓度的药物进行处理),每组设6个复孔(n=6)。
-
设空白组和药物组,药物组分别给予终浓度为200、100、50和25 μmol/L的雷公藤次碱药液进行处理。细胞分别培养24和48 h,实验结束4 h前加入10 μl CCK-8试剂,结束后以酶标仪于450 nm处检测吸光度(A)。
-
设空白组、模型组和药物组,药物组细胞给予LPS诱导后分别给予终浓度为100、50、25 μmol/L的药液进行处理。细胞培养24 h后取出,吸取细胞上清液,Griess试剂法检测上清中NO光密度值,ELISA试剂盒检测上清中IL-1β、TNF-α和IL-6的光密度值。
-
设空白组、模型组和药物组,药物组细胞给予LPS诱导后分别给予终浓度为100、50、25 μmol/L的药液进行处理。细胞培养24 h后取出,用4 ℃预冷的PBS液洗涤各孔两次,然后,吸弃PBS液,每孔中加入200 μl 4 ℃预冷的RIPA提取细胞蛋白,在13000 r/min下离心20 min,弃去上清,得各样本。用Bradford法对蛋白定量;使用细胞核/质提取试剂盒制备用于检测p65/NF-κB的胞质和核提取物。每孔加入20 μg蛋白上样,在SDS-PAGE凝胶中电泳,蛋白转移到PVDF膜上,用BSA封闭2 h。分别用相应一抗4 ℃孵育过夜,然后用与辣根过氧化物酶偶联的二抗(1∶1000)孵育2 h。最后用 ECL超敏发光液显影,于凝胶成像仪中曝光,使用 LAS成像软件对条带进行定量分析
-
实验数据的统计分析用(
$ \bar x \pm s $ )表示,采用单因素方差分析法进行统计学处理。应用Alpha软件处理Western Blot实验所得目标带的光密度值,并以各自的β-actin蛋白条带灰度值与目标条带灰度值的比值作为蛋白相对表达水平值。 -
图1显示了不同浓度雷公藤次碱对RAW264.7细胞的细胞毒性作用。给予雷公藤次碱(25~200 μmol/L)孵育24 h后,200 μmol/L雷公藤次碱可显著抑制RAW264.7细胞活性;孵育48 h后,50、100和200 μmol/L雷公藤次碱均可显著抑制RAW264.7细胞的活性。
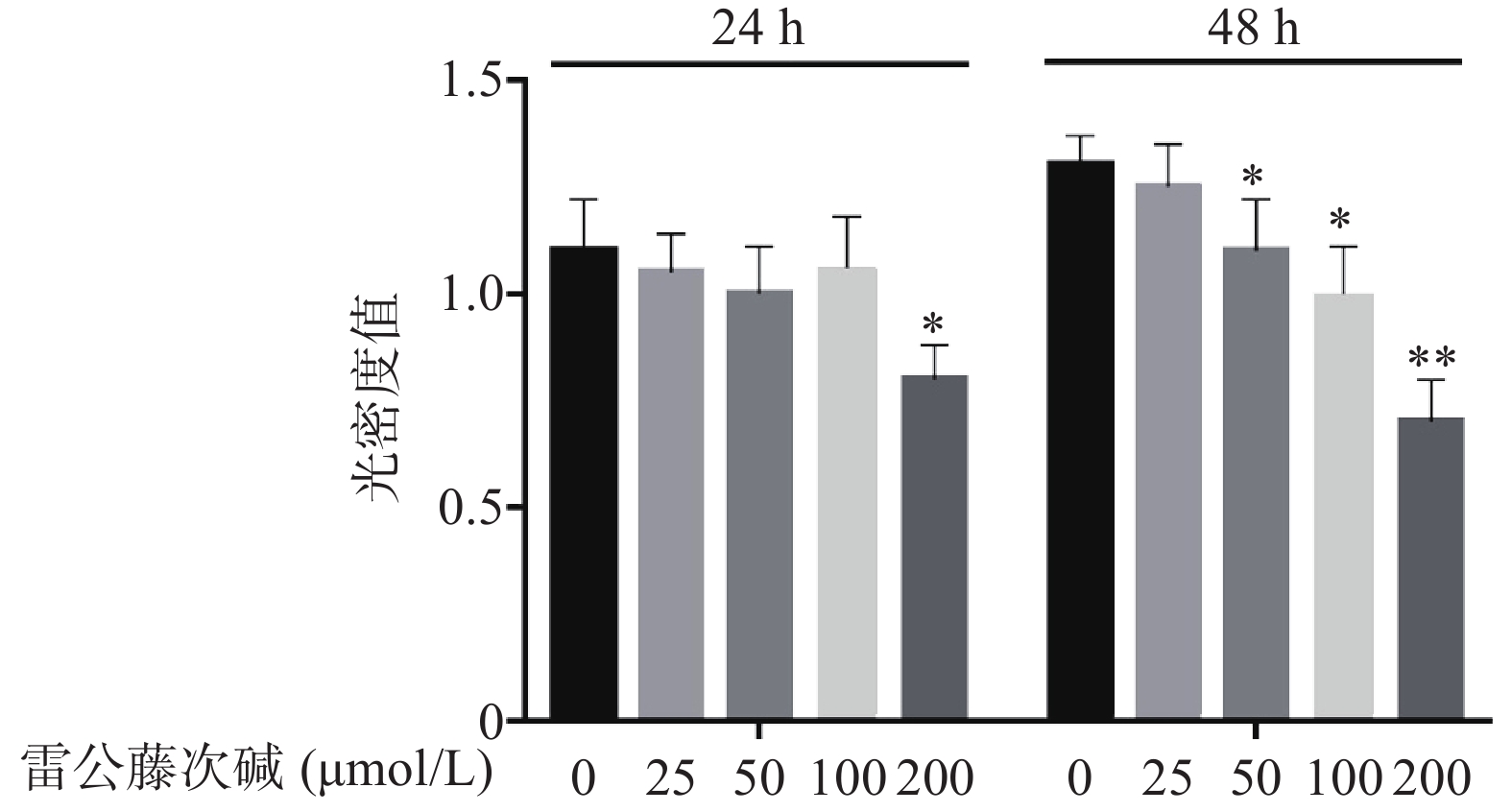
图 1 雷公藤次碱对RAW264.7细胞活力的影响
-
为了进一步研究雷公藤次碱体外抗炎活性,检测了雷公藤次碱对LPS刺激RAW264.7细胞分泌炎症因子的影响。结果显示(图2),给予LPS刺激后的RAW264.7细胞,NO、IL-1β、TNF-α和IL-6等促炎因子释放水平急剧增加(P<0.01),给予不同浓度雷公藤次碱(100、50和25 μmol/L)处理后,上述因子分泌水平呈剂量依赖性显著降低(P<0.01)。由此提示,雷公藤次碱的抗炎作用可能与抑制细胞释放NO、IL-1β、TNF-α和IL-6等促炎因子有关。
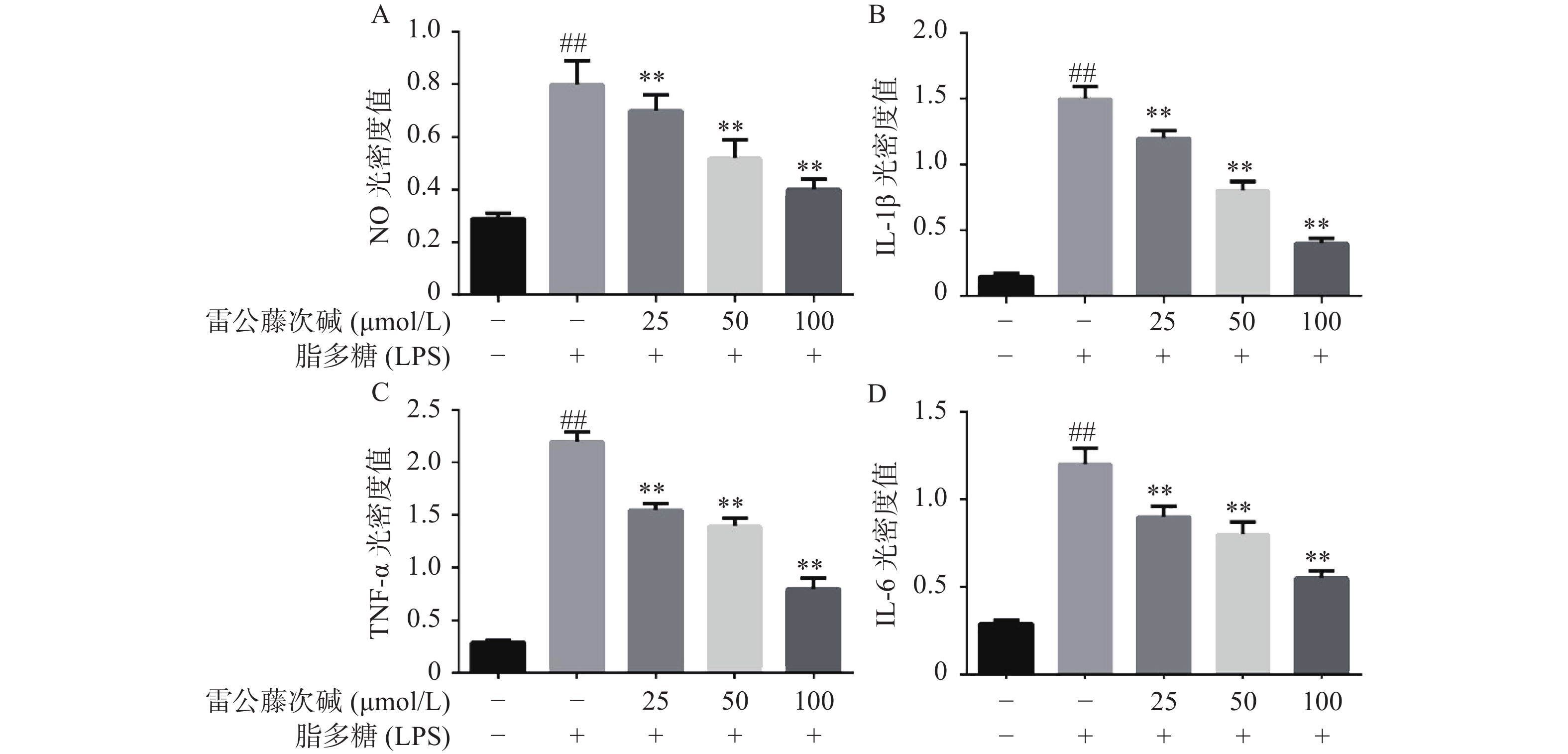
图 2 雷公藤次碱对LPS诱导RAW264.7细胞分泌炎症因子的影响
-
LPS是引发炎症的早期关键因素,可诱发多种细胞内信号事件,LPS-TLR4/MyD88信号通路可调节促炎基因表达,并控制炎症相关细胞因子的表达,其中IRAK及TRAF6是MyD88依赖性途径中的关键信号传导的分子。TLR4受体受LPS诱导后,可导致IRAK磷酸化从而脱离MyD88与TRAF6的结合体,使得游离的TRAF6活化后续信号转导途径[6]。为了评估雷公藤次碱对LPS激活TLR4/MyD88信号通路的影响,蛋白印迹法考察了雷公藤次碱对IRAK磷酸化和TRAF6表达的影响。结果显示(图3),与空白组相比,LPS刺激的模型组IRAK磷酸化和TRAF6表达明显增加(P<0.01);与模型组相比,雷公藤次碱各剂量组可显著抑制IRAK磷酸化和降低TRAF6表达(P<0.01)。

图 3 雷公藤次碱对TRAF6表达和IRAK磷酸化的表达的影响
-
NF-κB是MyD88信号通路中重要的转录因子[7]。我们考察了雷公藤次碱对LPS诱导的RAW264.7细胞中NF-κB活化的潜在影响。如图4所示,RAW264.7细胞LPS刺激后,细胞核内NF-κB p65水平增高而细胞质内NF-κB p65水平降低,说明NF-κB p65活化后由细胞质进入到细胞核内,而给予雷公藤次碱处理后,细胞质内NF-κB p65水平增高而细胞核内NF-κB p65水平降低,说明雷公藤次碱以浓度依赖的方式显著地阻止了NF-κB p65从细胞质转运到细胞核。 NF-κB p65易位进入核内与TLR4途径中的IκBα磷酸化有关。因此,我们考察了雷公藤次碱对NF-κB抑制蛋白IκBα磷酸化的影响。结果表明,雷公藤次碱以浓度依赖的方式显著抑制LPS诱导的IκBα磷酸化和降解。
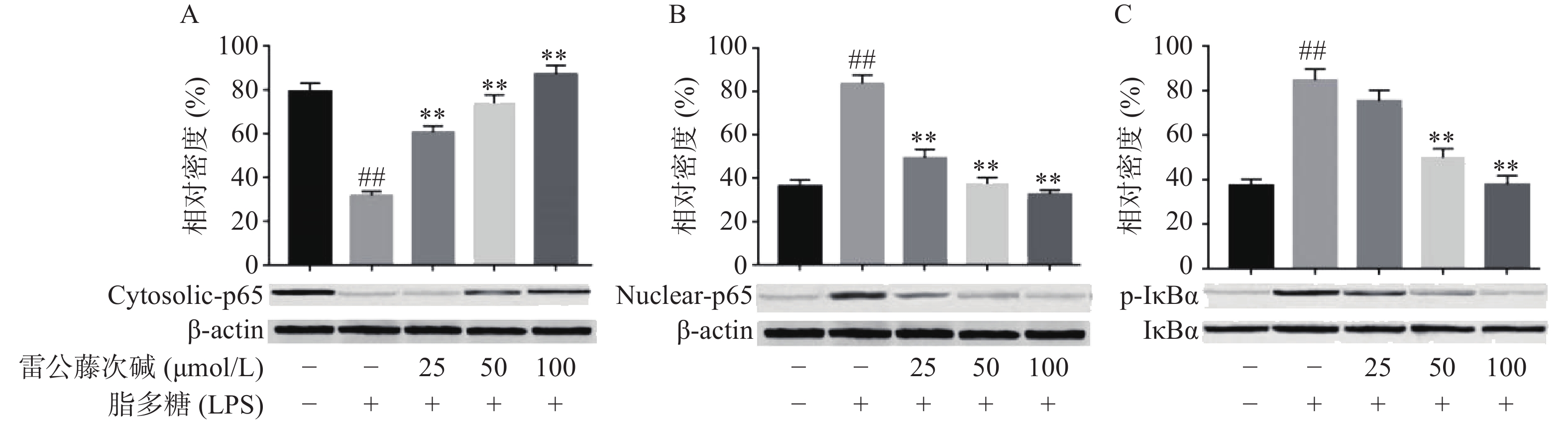
图 4 雷公藤次碱对IκBα磷酸化和NF-κB核转位的影响
-
MAPKs是信号从细胞表面传导到细胞核内部的重要传递者。为了进一步研究雷公藤次碱是否调节MAPKs,考察了其对LPS诱导的RAW264.7细胞中ERK、p38和JNK等MAPKs磷酸化的影响。如图5所示,LPS暴露可显著促进RAW264.7细胞中ERK、JNK和p38的磷酸化。给予雷公藤次碱可以显著抑制LPS诱导的ERK、p38和JNK的磷酸化,但并未影响ERK、p38和JNK的表达。上述结果表明,MAPKs的磷酸化可能参与了雷公藤次碱对RAW264.7细胞中LPS刺激炎症的抑制作用。
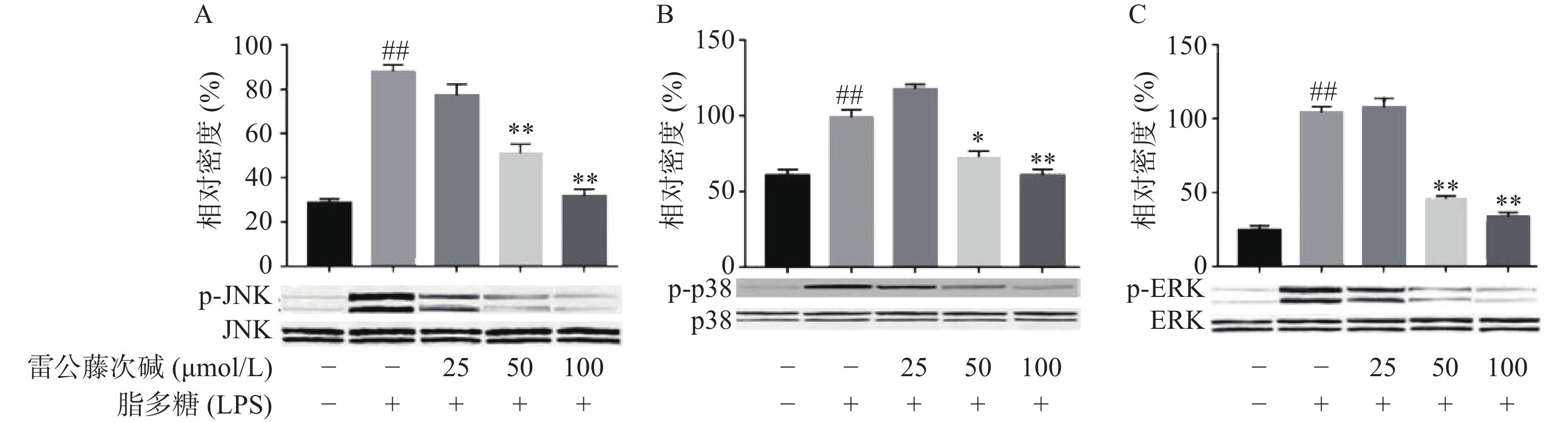
图 5 雷公藤次碱对p38, ERK和JNK MAPKs磷酸化的影响
-
雷公藤次碱是雷公藤的代表性生物碱成分之一,但是自从被分离以来,其生物学活性除了杀虫外,其他方面了解甚少[8]。本研究显示,雷公藤次碱对LPS诱导的RAW264.7细胞炎症模型显示出显著的抗炎活性。雷公藤次碱可减少LPS刺激引起的NO、IL-1β、TNF-α及IL-6等促炎因子的产生,并抑制LPS诱导的TLR4/MyD88通路中信号传导的分子IRAK磷酸化及TRAF6表达,从而影响MAPKs和NF-κB的激活,如图6所示。
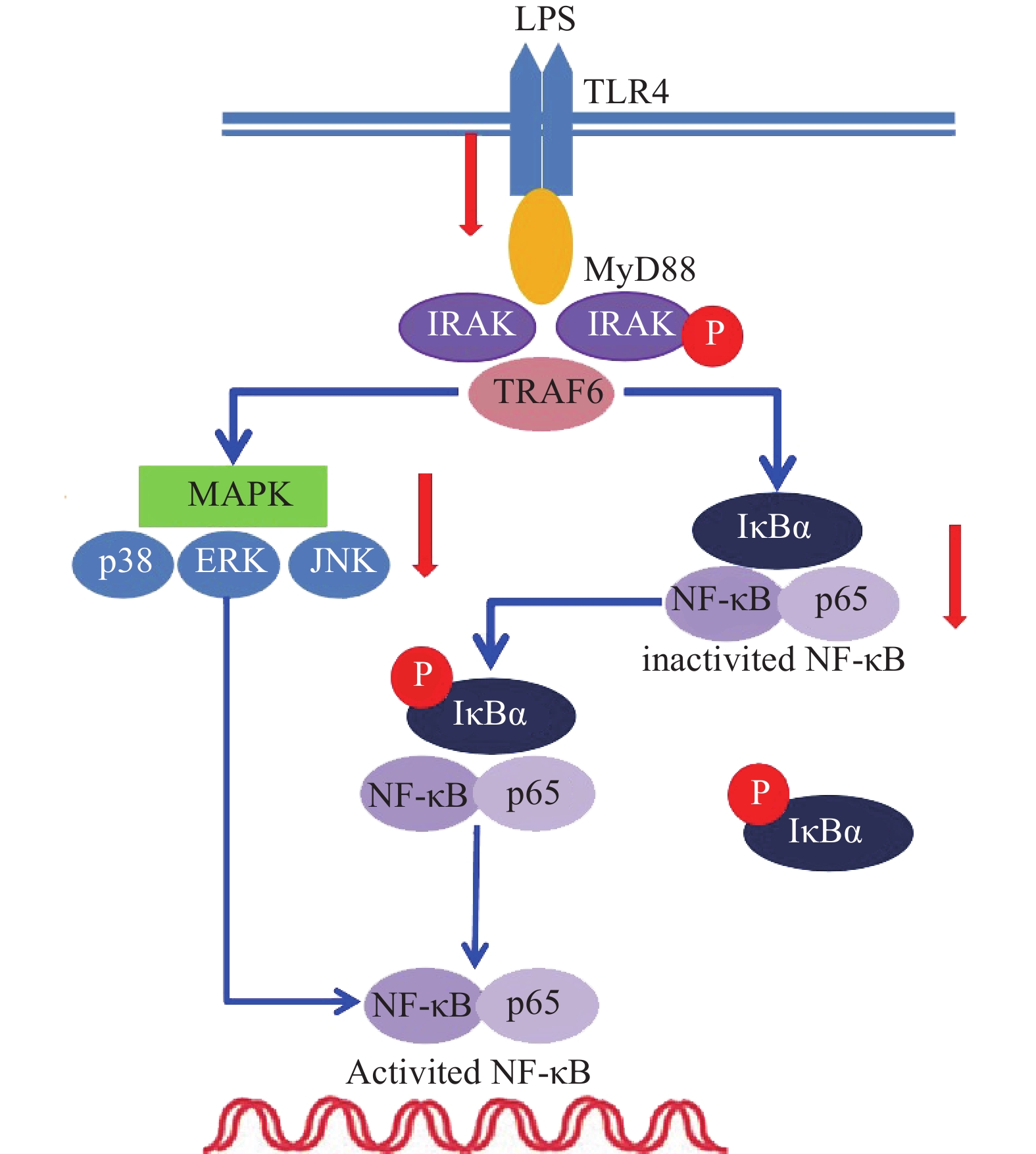
图 6 雷公藤次碱抗炎调控机制
已有证据显示,LPS可引发多种细胞内信号事件,包括导致TLR4,NF-κB及p38,ERK和JNK等丝裂素活化蛋白激酶(MAPKs)途径激活[9-10]。研究发现,LPS诱导后RAW264.7细胞Toll样受体4(TLR4)的表达明显增高,说明LPS是或可引导TLR4的配体与TLR4结合,二者结合后可启动细胞内一系列信号级联反应,最终诱导目的基因的表达。TLRs亚型中除TLR3外,均需要髓样分化因子88(MyD88)来激活下游释放信号分子的通路,这种途径成为MyD88依赖性途径。静息状态下,MyD88与IRAK及TRAF6形成细胞信号转导复合物,受到诱导后导致IRAK磷酸化,IRAK磷酸化后TRAF6从信号转导复合物中解离出来,TRAF6的活化可引起两条不同的信号转导途径,一条为MAPK家族(包括p38、JNK),另一条为活化MP3K(mitogen activated protein kinase kinase kinase)家族成员NIK(NF-κB inducing kinase),后者的磷酸化激活IκB激酶,导致IκB的磷酸化而使NF-κB/IκB复合物解离,NF-κB由此活化转位进入细胞核[11]。在本研究中,数据显示雷公藤次碱可以抑制IRAK磷酸化和TRAF6的表达。
NF-κB是一种多效性的转录因子,参与调控炎症反应、免疫、肿瘤等相关的细胞因子、趋化因子、黏附分子、炎症介质等的转录过程。IκB是其抑制蛋白,正常情况下二者结合而使NF-κB处于失活状态,IκBα是IκB的亚分子,受到LPS刺激,IκBα磷酸化增加,从而导致游离NF-κB p65释放并从细胞质转移至细胞核,与DNA上的相应位点结合,导致特定靶基因(如TNF-α、IL-1β和IL-6)的转录表达[12-13]。在本研究中,数据显示雷公藤次碱可抑制IκBα磷酸化,并以浓度依赖的方式阻止NF-κB p65由细胞质进入细胞核,从而阻止炎症相关因子的转录表达。
此外,NF-κB的易位也受ERK、p38和JNK等丝裂素活化蛋白激酶途径调节,该途径可控制炎症反应过程中细胞因子的合成和释放。丝裂素活化蛋白激酶激活后,细胞质或细胞核中的转录因子又被激活,从而触发引起生物学反应的靶基因的表达,包括促炎性介质的表达。本研究显示,LPS诱导的RAW264.7细胞中,雷公藤次碱可显著抑制ERK、JNK和p38的磷酸化。
Wilforine inhibits LPS-induced inflammatory response in RAW264.7 cells by regulating the TLR4/MyD88/TRAF6 signaling pathway
-
摘要:
目的 探讨雷公藤次碱抗炎活性及其作用机制。 方法 用细胞计数盒-8 (CCK-8)法考察雷公藤次碱对小鼠单核巨噬细胞白血病细胞RAW 264.7 增殖活性的影响,用酶联免疫吸附法(ELISA)检测雷公藤次碱对脂多糖(LPS)诱导的RAW264.7细胞分泌细胞因子一氧化氮(NO)、白细胞介素-1β(IL-1β)、 肿瘤坏死因子α(TNF-α)和白细胞介素6(IL-6)的影响,用免疫印迹法考察雷公藤次碱对白介素受体相关激酶(IRAK)、肿瘤坏死因子受体相关蛋白6(TRAF6)、核因子κB抑制因子α(IκBα)、核因子κB(NF-κB p65)、分裂原激活的蛋白激酶p38(p38)、c-Jun氨基末端激酶(JNK)、细胞外调节蛋白激酶(ERK)在LPS刺激的RAW264.7细胞中的表达及其磷酸化的影响。 结果 雷公藤次碱在25、50、100 μmol/L浓度下对RAW264.7细胞无显著毒性,并可显著抑制细胞因子NO、IL-1β、TNF-α和IL-6含量。免疫印迹法检测结果显示雷公藤次碱可显著抑制IRAK及TRAF6的表达;显著抑制ERK、P38和JNK的磷酸化;抑制IκBα的降解,降低NF-κB p65的核转运水平。 结论 雷公藤次碱具有体外抗炎活性,其作用机制可能介导TLR4/MyD88/TRAF6信号通路。 Abstract:Objective To investigate the anti-inflammatory effect and mechanism of wilforine. Methods Anti-inflammatory activity of wilforine was investigated in LPS-induced RAW264.7 cells. The cytokines production of RAW264.7 cells was analyzed by ELISA assay and the cell viability was assessed by CCK-8 method. The expression of TRAF6, the phosphorylation of IRAK, p38, ERK and JNK, the degradation of inhibitory κBα (IκBα) and the nuclear translocation of NF-κB p65 were further investigated by western blot. Results Triptolide had no significant toxicity to RAW264.7 cells at concentrations of 25, 50 and 100 μmol/L. and could significantly inhibit the contents of cytokines NO, IL-1β, TNF-α and IL-6. Wilforine significantly decreased the expression of TRAF6 and phosphorylation of IRAK, and inhibited the phosphorylation of ERK, p38, and JNK and degradation of IκBα, and reduced the level of nuclear translocation of NF-κB p65. Conclusion The anti-inflammatory activity of wilforine of LPS-induced RAW264.7 cells is probably via TLR4/MyD88/TRAF6 signaling pathway. -
Key words:
- Tripterygium wilfordii /
- wilforine /
- anti-inflammation /
- TLR4 /
- MyD88 /
- NF-κB
-
冠状病毒(coronavirus,CoVs),在电子显微镜下呈“冠状”或冠状形态,因而命名为冠状病毒,可感染人类并容易引起急/慢性呼吸道系统疾病[1]。冠状病毒科分为α、β、γ和δ 4个属[2],已知感染人的冠状病毒共有7种,分别是HCoV-229E、HCoV-OC43、HCoV-NL63、HCoV-HKU1、SARS-CoV,MERS-CoV和SARS-CoV-2,其中,前4种病毒致病性较低,一般仅引起类似普通感冒的轻度感染,后3种对人类健康产生严重威胁,具有较高的致死率,分别是2003年出现的冠状病毒(severe acute respiratory syndrome coronavirus,SARS-CoV)感染的严重急性呼吸道综合征、2012年在中东地区出现高致病性冠状病毒(Middle East respiratory syndrome coronavirus,MERS-CoV)感染的中东呼吸综合征和2019年出现的一种新型冠状病毒(severe acute respiratory syndrome coronavirus 2,SARS-CoV-2)感染引起的肺炎(Corona Virus Disease 2019,COVID-19)[3]。
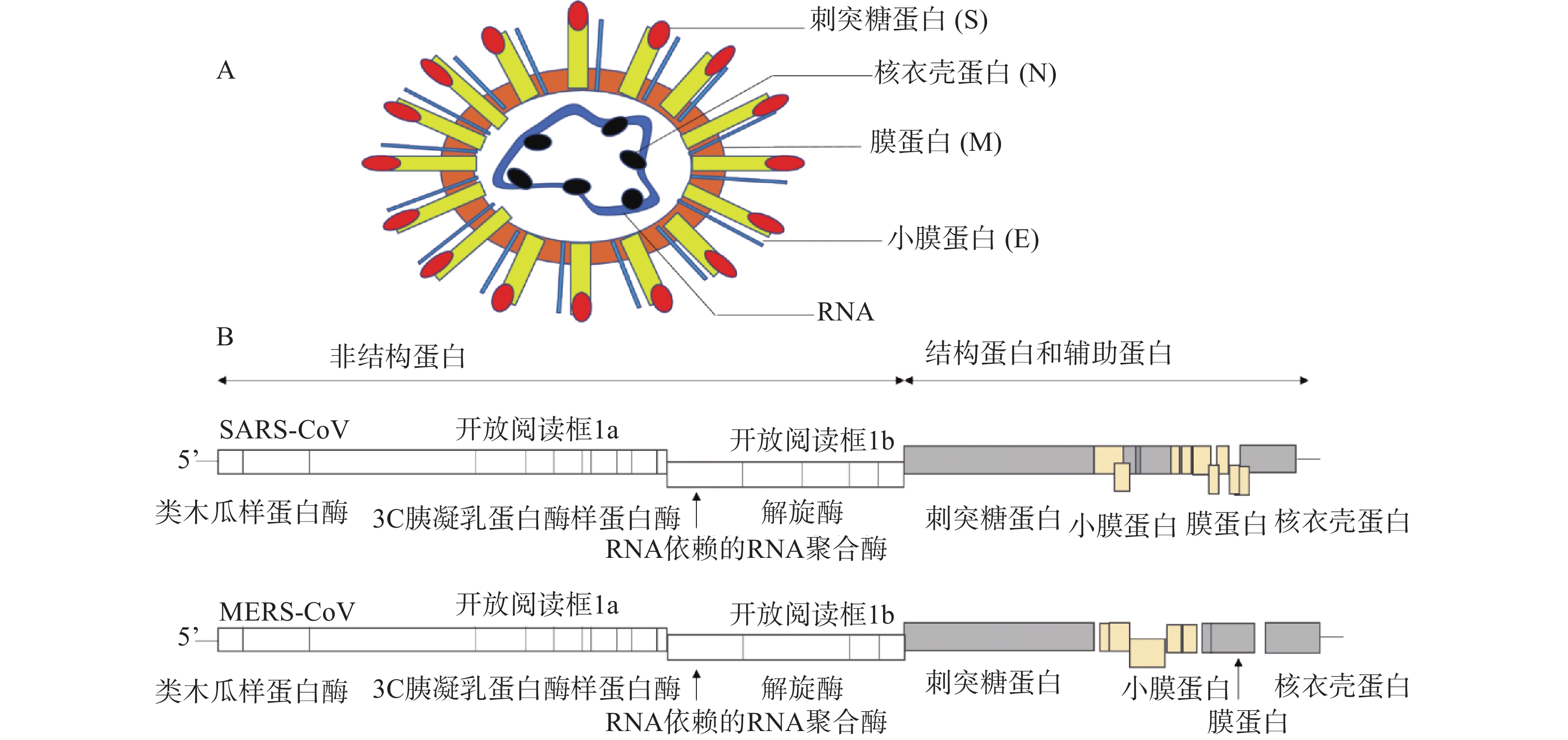
1. 冠状病毒及其药物靶点
冠状病毒是一种包膜病毒,具有直径为120~160 nm的圆形或椭圆形病毒体[4]。冠状病毒粒子外包裹着脂肪膜,膜表面有3种糖蛋白,分别是刺突糖蛋白(spike glycoprotein,S 蛋白)、膜蛋白(membrane protein,M 蛋白)和小膜蛋白(envelope protein,E 蛋白)。其中,S蛋白是在病毒体表面上形成配体的I型糖蛋白,介导与宿主受体的连接,由S1和S2两个亚基组成;M蛋白是一种前蛋白,主要参与病毒的装配过程;E蛋白是一种高度疏水的蛋白,可促进病毒的组装和释放。病毒的基因组RNA与核衣壳蛋白(nucleocapsid,N蛋白)在病毒膜内复合形成螺旋衣壳(图1A)。
冠状病毒RNA基因组是不分段的单股正链RNA,大小为26~32 kb,是RNA病毒中基因组最大的病毒[5]。冠状病毒的基因顺序一致,为5'-复制酶-S-E-M-N-3',其中,RNA 5'端为被封端,具有甲基化的帽状结构,3'端被聚腺苷酸化,具有poly(A)尾巴,具有传染性。从5'端开始,基因组长度的2/3(长度:20~22 kb)区域由两个重叠的开放阅读框(open reading frame)1a和1b组成,共同起着病毒RNA聚合酶(Pol)的作用,编码翻译成两个多肽,pp1a和pp1ab[6]。这些多聚蛋白酶包括:类木瓜样蛋白酶(papain-like protease,PLpro)、3C胰凝乳蛋白酶样蛋白酶(3C chymotrypsin like protease, 3CLpro,也称为主蛋白酶,Mpro)、RNA依赖的RNA聚合酶(RNA-dependent RNA polymerase, RdRp)和解旋酶(helcase,Hel)等[7]。对于所有冠状病毒,结构蛋白编码在3′端,占基因组的1/3,按S-E-M-N顺序排列(图1B)。
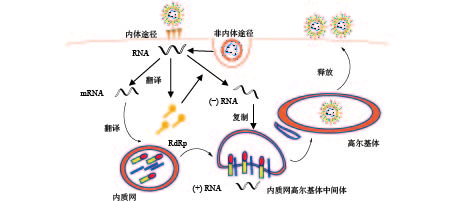
冠状病毒通过S蛋白附着在特定的细胞受体上,S1亚基和同源受体之间的特异性相互作用会触发S2亚基的构象变化,导致病毒包膜和细胞膜融合,并释放核衣壳进入细胞质;病毒在组织蛋白酶介导下与细胞表面受体结合以内体途径或细胞表面非内体途径进入宿主细胞,低pH和pH依赖性的半胱氨酸蛋白酶、组织蛋白酶可以促进冠状病毒通过内体途径进入宿主细胞[8]。病毒进入细胞后,正链RNA翻译产生负链RNA聚合酶前体蛋白,通过蛋白质水解过程产生RNA依赖性RNA聚合酶(RdRp);在RNA聚合酶的作用下,生成全长的反义负链模板。从负链亚基因组模板中合成亚基因组mRNAs;亚基因组mRNAs的翻译会产生病毒结构蛋白,在内质网(endoplasmic reticulum, ER)中进行病毒膜蛋白装配,并形成小泡运出。在内质网高尔基体中间体(ER-Golgi intermediate compartment, ERGIC)中被衣壳蛋白包裹的正链RNA与膜蛋白识别,小泡内卷,包住RNA。病毒通过高尔基体(golgi apparatus, GA)释放囊泡,囊泡与质膜融合释放病毒,病毒继续感染其他细胞(图2)。冠状病毒RNA在复制过程中,因纠正其错误复制的酶活性较低,很容易出现病毒错误复制,导致病毒变异株的出现。
冠状病毒的生命周期是一个复杂、连续、多步的过程,病毒配体S蛋白、与病毒转录复制相关的RdRp、PLpro和3CLpro等多种蛋白质和酶发挥关键作用,是治疗冠状病毒感染疾病的关键药物靶标,以它们为靶点,国内外研究者发现了一系列的小分子抑制剂[9-22],现分述如下。
2. 冠状病毒小分子抑制剂
2.1 SARS-CoV小分子抑制剂
2.1.1 肽类小分子抑制剂
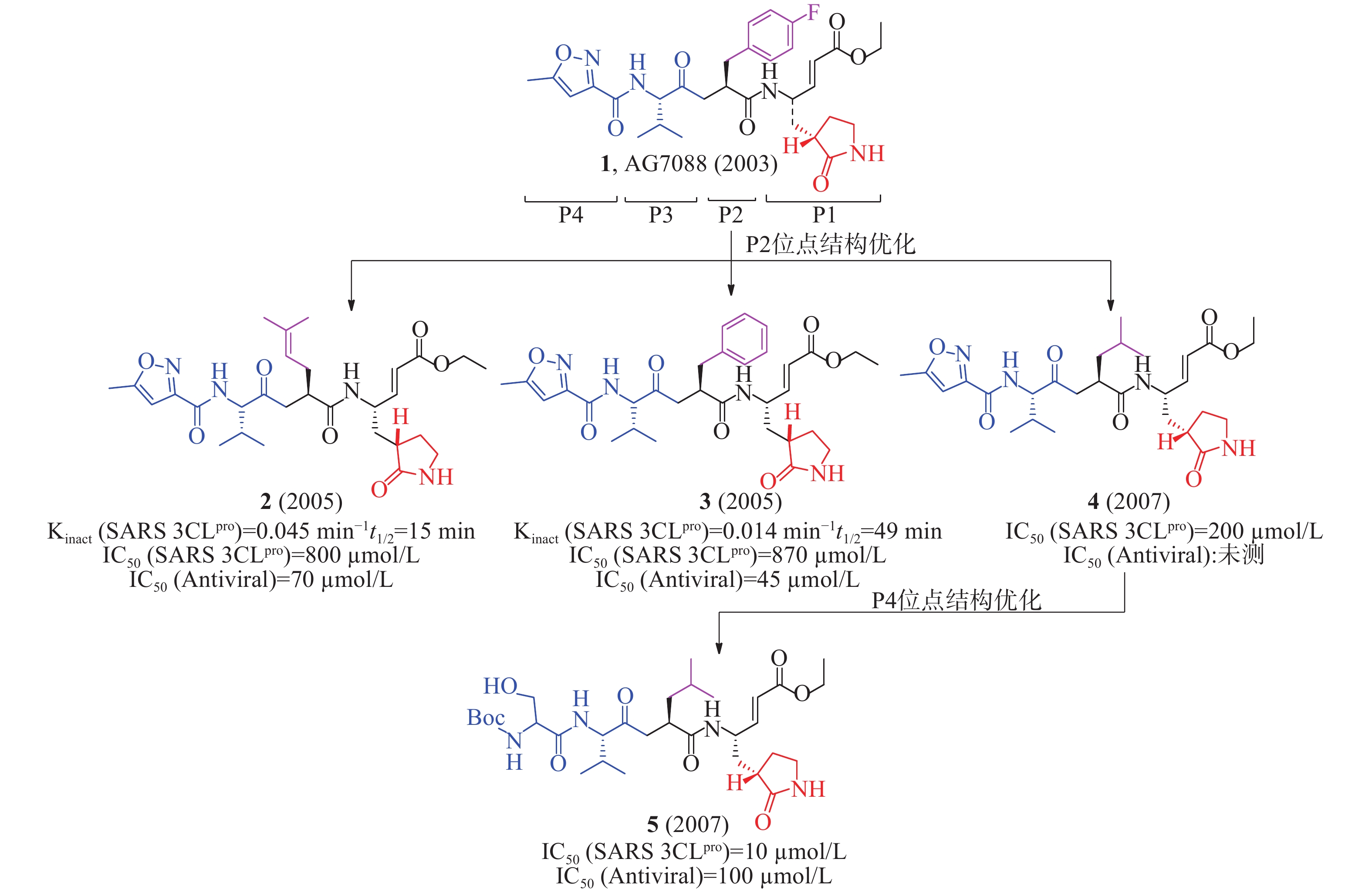
2003年,Hilgenfeld等报道了人冠状病毒(229E株)3CLpro(HCoV 229E 3CLpro)和猪传染性胃肠炎病毒(transmissible gastroenteritis virus, TGEV) 3CLpro的晶体结构,为抗冠状病毒药物研究提供了重要的结构生物学基础。Hilgenfeld等以此同源模建了SARS-CoV 3CLpro的三维结构,通过分子对接发现多肽类似物1(AG7088, rupintrivir,图3)可能对SARS-CoV 3CLpro具有良好的抑制活性[11]。该化合物原是一种鼻病毒3Cpro抑制剂,用于治疗普通感冒,但口服生物利用度低,需鼻腔给药,临床研究已终止[12]。
2005年,Mesecar等确定AG7088与SARS 3CLpro结合的晶体结构,为后续结构优化和构效关系研究提供了重要的结构生物学基础。根据AG7088与SARS-3CLpro的结构模式,AG7088可划分为P1~P4四个区域。以此为模板对P2位点进行改造,合成了一系列多肽类小分子抑制剂。用荧光共振能量转移法(fluorescence resonance energy transfer, FRET)酶水平活性研究表明,含有2-甲基丙烯基基团的化合物2(Kinact= 0.045/min,t1/2= 15 min, 图3)活性优于含有苯基基团的化合物3(Kinact= 0.014/min,t1/2= 49 min),提示P2位点对应SARS 3CLpro的空腔可以容纳较小侧链残基[13]。以此为模板对P4位点再进行改造,又合成了一系列多肽类小分子抑制剂。化合物4的IC50值为200 μmol/L,但抗病毒活性丧失;随后,以化合物4(图3)为先导,在P4位点优化获得的化合物5[IC50=10 μmol/L,IC50(antiviral)=100 μmol/L]比化合物4[IC50= 200 μmol/L,IC50(antiviral):未测]对SARS 3CLpro抑制活性和抗病毒活性更好。该化合物P4位点从原有的5-甲基异恶唑-3-基团变成1-[(叔丁氧羰基)氨基]-2-羟基乙烷-1-基团,易与SARS 3CLpro的谷氨酸(166)残基形成更多的氢键相互作用,有利于提高活性[14]。
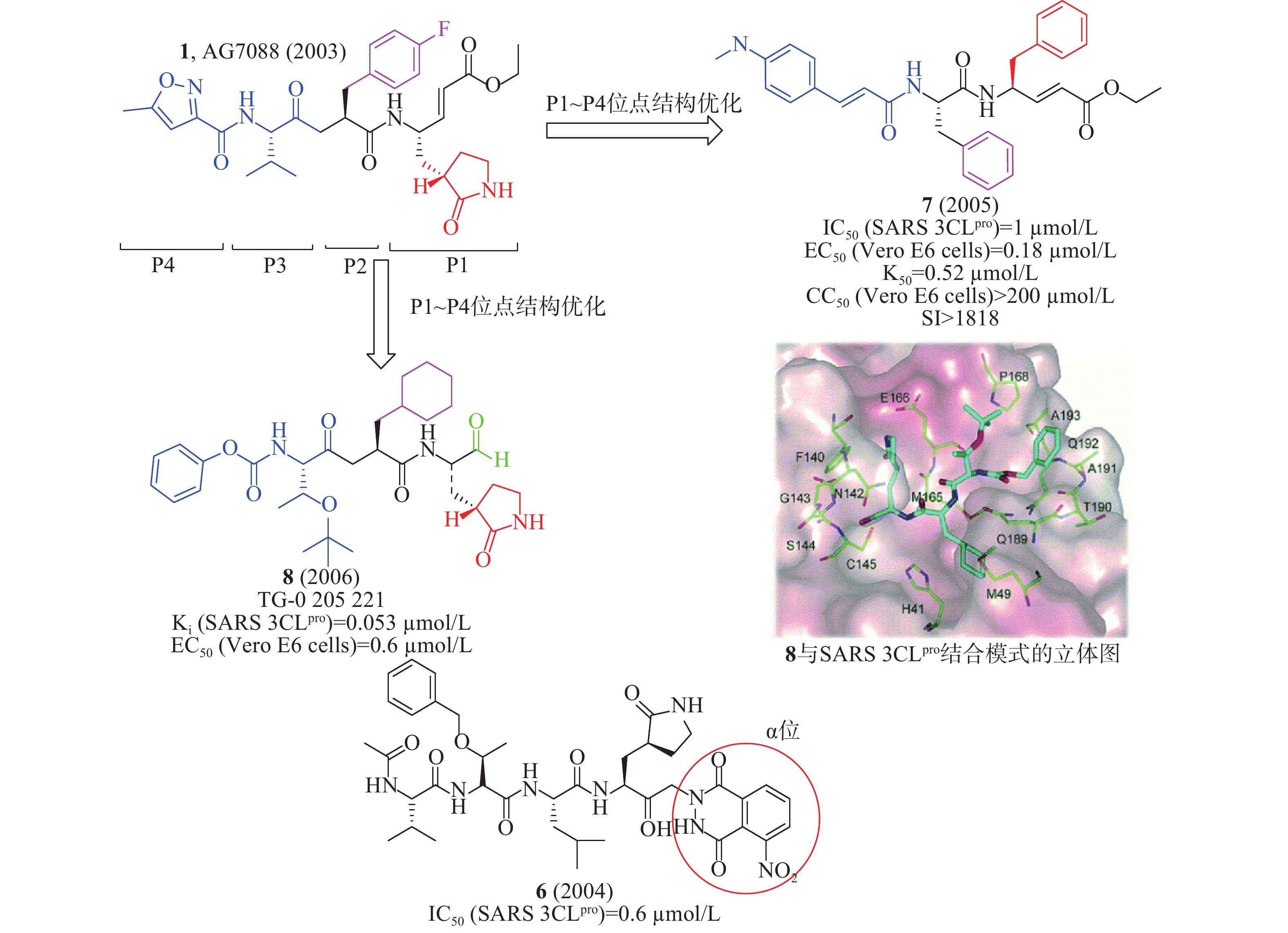
2004年,Vederas等合成了一系列在α位带有邻苯二甲酰肼基团的酮-谷氨酰胺类似物,并测试其抑制抗SARS-CoV 3CLpro的活性。在酶水平系统构效关系表明:将三肽(Ac-Val-Thr-Leu)附着在谷氨酰胺上可产生更好的抑制剂6(IC50= 0.60 µmol/L,图4)[15]。2005年,Wong等以1为先导化合物,对其P1~P4位点优化设计,合成了一系列衍生物,其中,化合物7(图4)在酶水平对SARS-CoV 3CLpro的IC50值为1 µmol/L、Ki(inhibition constant)值为0.52 µmol/L,在感染了SARS-CoV的Vero E6细胞中测得的EC50值为0.11 µmol/L和CC50值>200 µmol/L,选择性指数(SI,selectivity index, CC50/EC50)>1 818[16]。
2006年,Hsu等发现了化合物8(Ki= 53 nmol/L,图4),在SARS-CoV感染的Vero E6细胞EC50值为0.6 µmol/L。通过解析化合物8与SARS 3CLpro结合的晶体结构,化合物8 P1位点醛基的羰基基团与SARS 3CLpro的Cys145共价结合形成C-S共价键、与SARS 3CLpro的Gly143和Cys145形成两个N-H氢键、P1位点五元环内酰胺基团与SARS 3CLpro的His163、Phe140、Glu166形成3个氢键,P2和P3位点的N-H键分别与Gln189和Glu166的羰基氧原子形成氢键、P3位点的羰基氧原子与SARS 3CLpro的Glu166的N-H键形成氢键,这些共价键作用和氢键作用使化合物8和SARS 3CLpro紧密结合[17]。
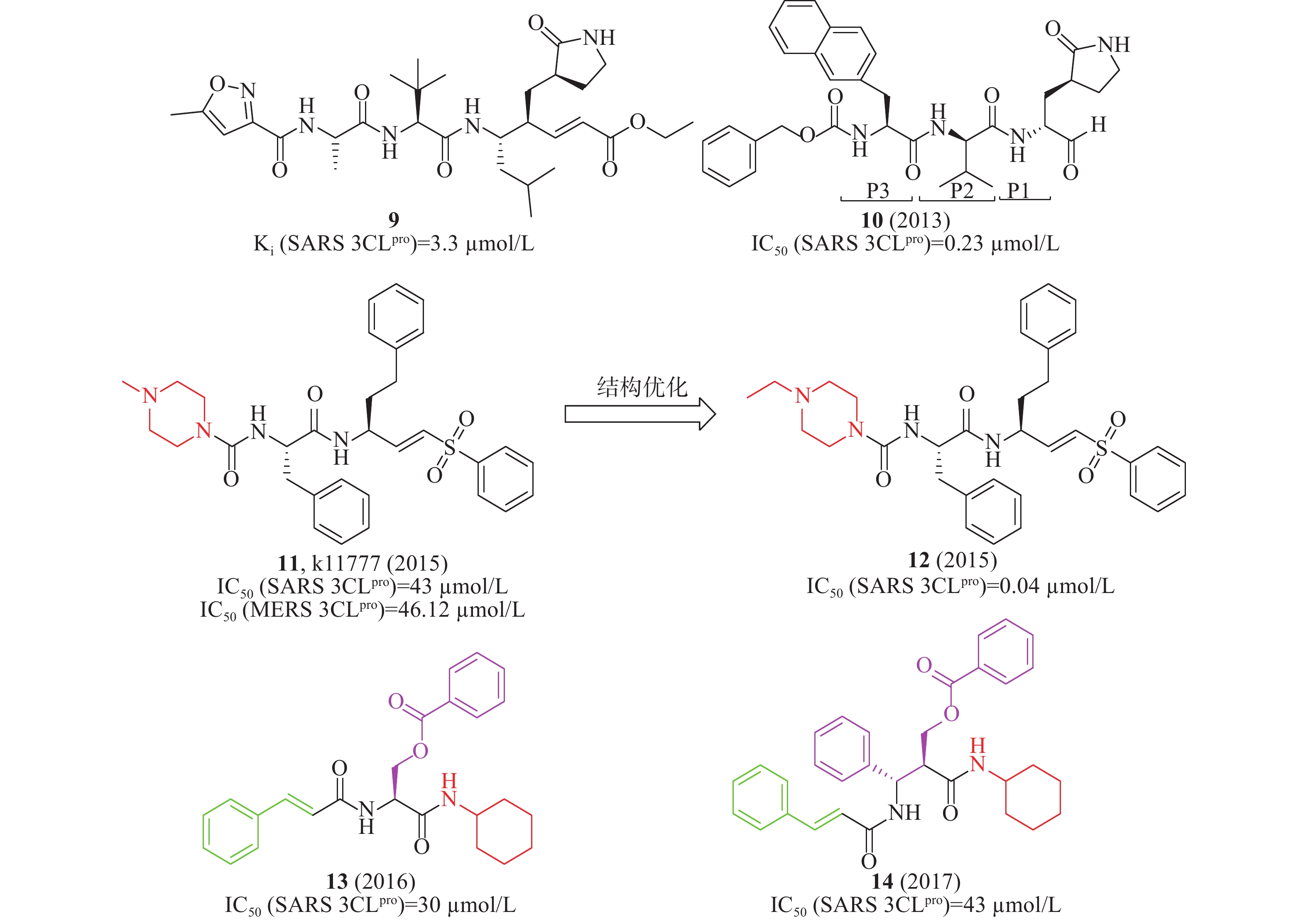
此外,国内外还报道了一系列多肽或多肽类似物作为SARS-CoV 3CLpro抑制剂。2008年,Rao等解析了SARS-CoV 3CLpro的突变体H41A晶体结构,发现了化合物9(Ki= 3.3 µmol/L,图5)在酶水平上对SARS-CoV 3CLpro具有抑制作用,可以作为SARS-CoV 3CLpro小分子抑制剂[18]。2013年,Hua等通过基于片段的药物设计策略设计合成了一系列3CLpro抑制剂,当P1位置为谷氨酰胺,P2位置为亮氨酸,P3位置为萘丙氨酸残基时,化合物10(图5)在酶水平对SARS-CoV 3CLpro的IC50值为0.23 µmol/L [19]。2015年,Simmons等筛选含有2100个化合物库,发现了化合物11(k11777,图5)在酶水平(IC50= 0.68 nmol/L)对SARS-CoV 3CLpro的活性最好,对其优化将哌嗪环上的甲基换成乙基时,在酶水平化合物12(图5)的IC50值为0.04 nmol/L [20]。2011年,Akaji等报道了丝氨酸衍生物13(图5)在酶水平对SARS-CoV 3CLpro的抑制活性较好,IC50值为30 µmol/L [21],进一步优化获得的苯异丝氨酸衍生物14(IC50= 43 µmol/L, 图5)比化合物13(IC50= 30 µmol/L)的活性低[22]。
2.1.2 非肽类小分子抑制剂
(1)天然产物
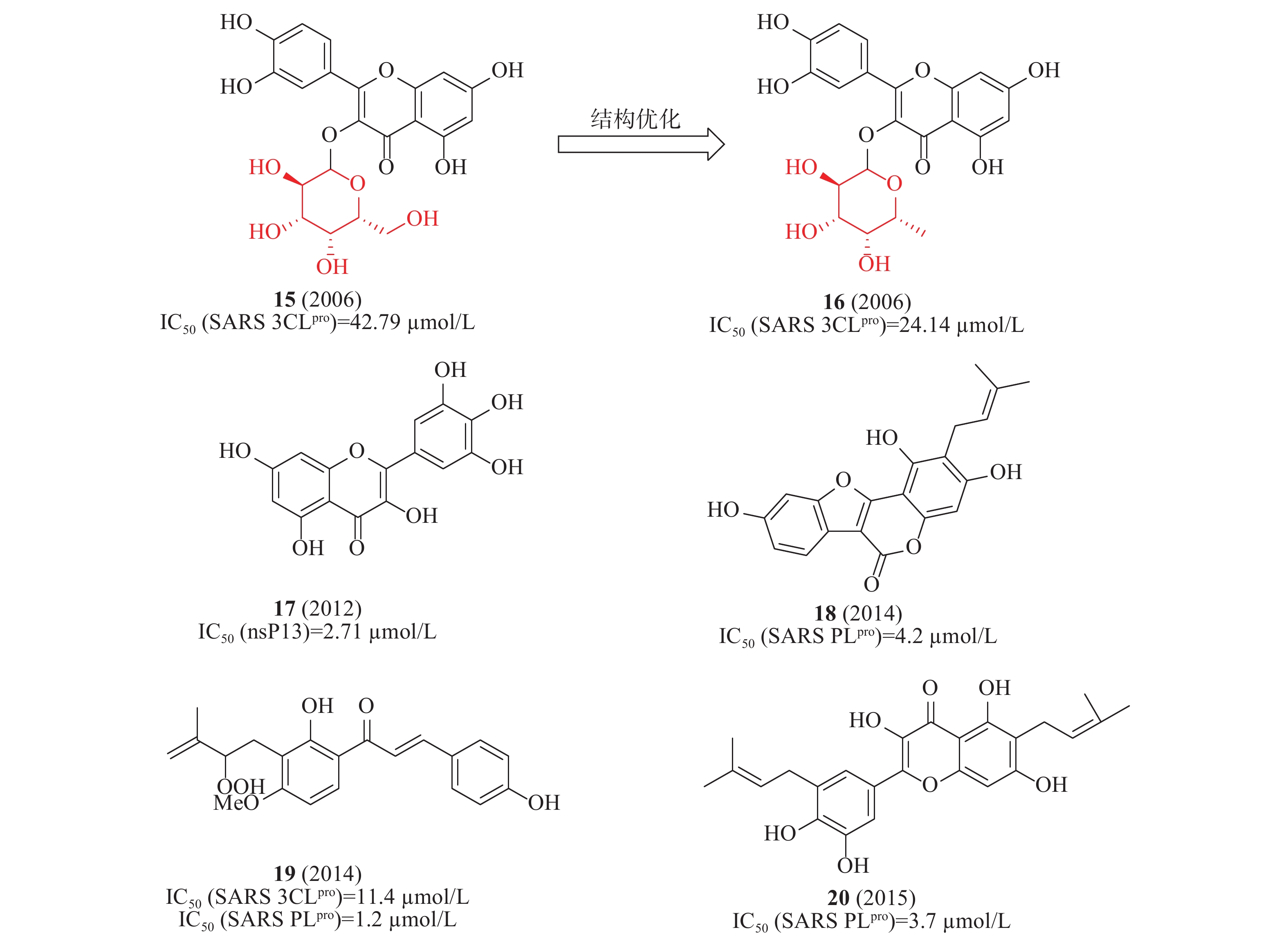
2006年,蒋华良等首次报道一种天然化合物15(槲皮素-3-β-半乳糖苷,图6)具有SARS-CoV 3CLpro抑制活性(IC50=42.79 μmol/L)。在分子对接模拟的基础上,由15为模板设计合成了8种衍生物,构效关系研究发现:①去除了槲皮素部分的7-羟基,活性降低;7-羟基上引入一个大的糖基取代基对活性影响不大;②糖基乙酰化使得活性丧失;③用其他糖取代半乳糖不影响活性。其中,化合物16(图6)活性较好,IC50为24.14 μmol/L[23]。2012年,Jeong等运用FRET法研究了64种天然化合物对SARS解旋酶的抑制作用。化合物17(杨梅素, 图6)通过影响ATP酶活性而非解旋酶活性,间接抑制了SARS-CoV解旋酶蛋白的表达,在酶水平对SARS-CoV病毒活性IC50值为2.71 μmol/L[24]。2014年,Park等发现补骨脂种子的乙醇提取物具有抗SARS-CoV PLpro活性,在酶水平IC50值为15 μmol/L。进一步分离获得6种芳香族化合物,它们均以剂量依赖性方式抑制PLpro,其中化合物18活性最佳(图6)在酶水平对SARS-CoV PLpro的IC50为4.2 μmol/L[25]。2015年,Ryu等发现含有羟基的查尔酮化合物19(图6)对3CLpro和PLpro的抑制IC50值分别为11.4和1.2 μmol/L[26]。2017年,Lee等从布鲁氏假单胞菌提取的多酚类化合物对3CLpro和PLpro具有抑制活性,多酚类化合物对PLpro的活性强于3CLpro;其中,化合物20(图6)对PLpro抑制活性最佳,IC50值为3.7 μmol/L[27]。
(2)三氮唑类
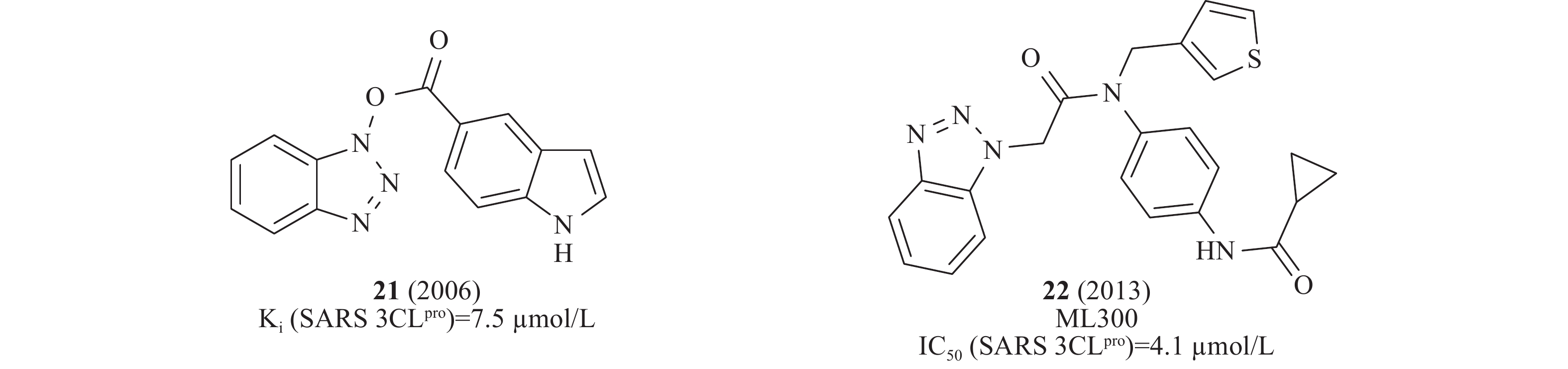
2016年,Wong等采用微滴定板组合反应策略修饰先导化合物,从中发现一类稳定的苯并三唑酯SARS-CoV 3CLpro抑制剂,动力学和质谱分析表明,该类化合物通过酰化修饰3CLpro的活性位点Cys145发挥作用,其中,化合物21(图7)在酶水平活性最佳,Ki为7.5 nmol/L[28]。此前,Stauffer等筛选NIH分子探针库(Molecular Libraries Probe Production Centers Network, MLPCN)发现了类似结构的SARS-CoV 3CLpro抑制剂,化合物22(图7)在酶水平对SARS-CoV 3CLpro的IC50为4.1 μmol/L[29]。
(3)咪唑类
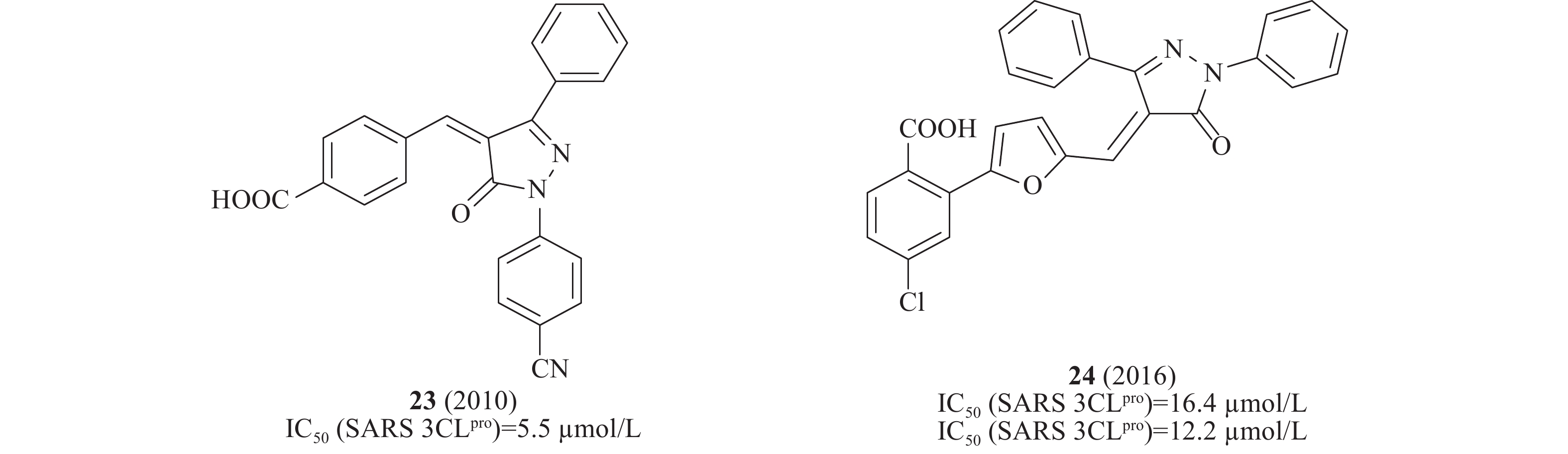
2010年,Liang等设计一系列吡唑啉酮类化合物作为SARS-CoV 3CLpro抑制剂,其中,化合物中23(图8)在酶水平对SARS-CoV 3CLpro表现出了较强的抑制作用,IC50为5.5 μmol/L[30]。2016年,Liang等通过“老药新用”的方法筛选神经氨酸酶(neuraminidase)对SARS-CoV的作用,化合物24(图8)在酶水平对SARS-CoV 3CLpro 的IC50为16.4 μmol/L[31]。
(4)萘环类
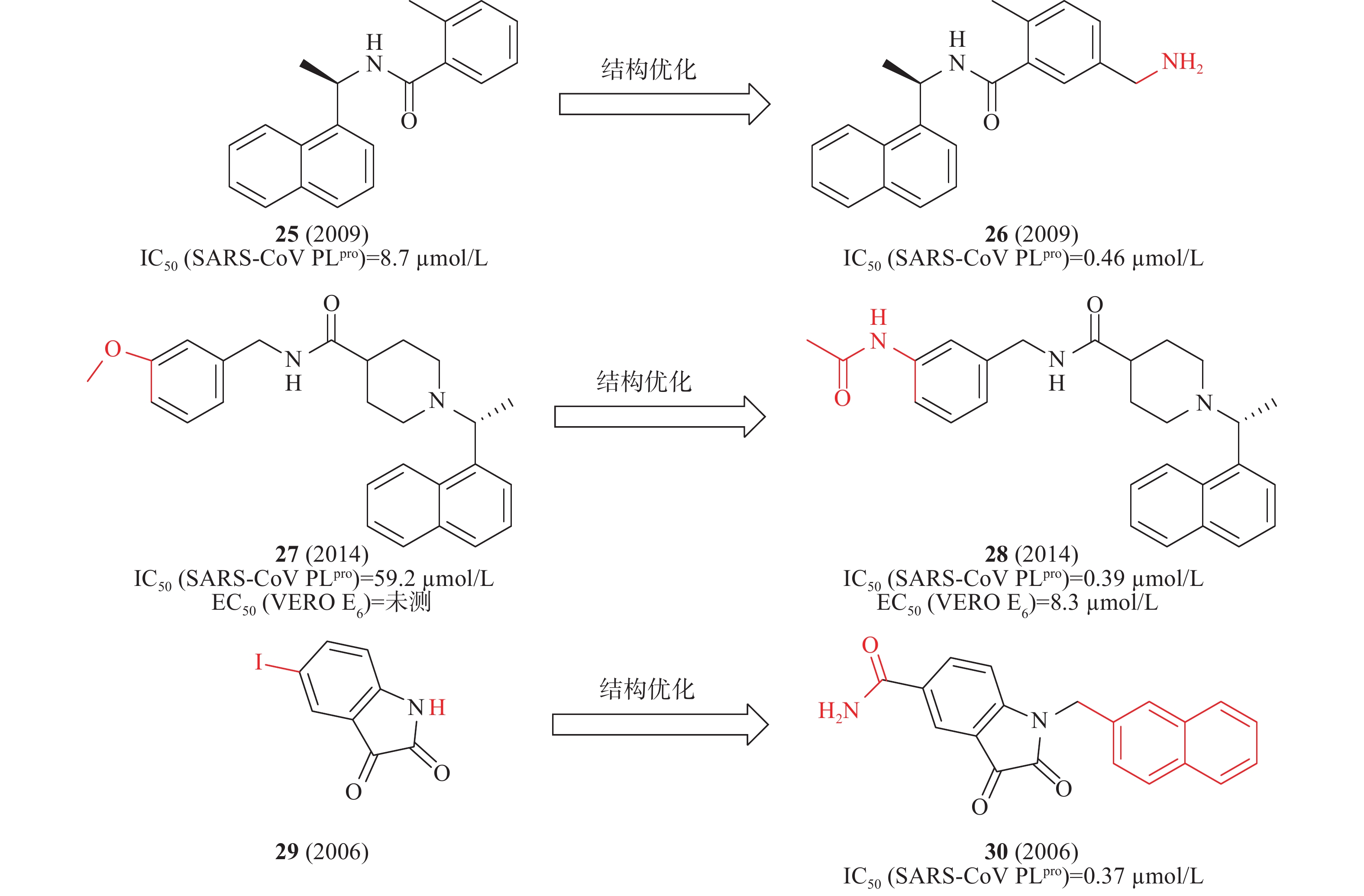
2014年,Mesecar等对多种化学文库的高通量筛选,确定化合物25在酶水平具有SARS-CoV PLpro抑制活性IC50为8.7μmol/L。以此为先导化合物,开展结构优化和构效关系研究,发现化合物26(图9)中引入乙胺基基团与SARS-CoV PLpro的Gln270、Tyr269形成氢键,酶水平提高了20倍(IC50=0.46 μmol/L),对SARS-CoV感染的Vero E6细胞的抗病毒活性为EC50=6 μmol/L[32]。2010年,Mesecar等通过对多种化学文库进行高通量筛选,鉴定了一种新的先导化合物27(图9),在酶水平对SARS-CoV PLpro抑制活性IC50为59.2 μmol/L,在SARS-CoV感染的Vero E6细胞中无抗病毒活性。以27为先导化合物合成一系列类似物和进行构效关系研究,将甲氧基替换为乙酰胺基优化得到化合物28(图9),28中的乙酰胺基基团与SARS-CoV PLpro的Tyr269形成氢键,28与SARS-CoV PLpro结合更加紧密,在酶水平(IC50=0.39 μmol/L)和抑制SARS-CoV感染的Vero E6细胞(EC50=8.3 μmol/L)方面同样有效[33]。2006年,来鲁华等以化合物29(图9)为模板,集中改变N-1和C-5位置取代基合成了一系列靛红类衍生物,研究了其构效关系。当N-1位置为β-萘亚甲基,C-5位置为甲酰胺时,化合物30在酶水平(图9)表现出明显的抑制作用,IC50为0.37 μmol/L[34]。
(5)喹啉类
2004年,Keyaerts等检测了抗疟药氯喹对感染SARS-CoV 的Vero E6细胞的抗病毒潜力。结果表明,化合物31(图10)的IC50值为8.8 μmol/L[35]。2005年,Nichol等发现化合物31(氯喹)对SARS-CoV(ED50为4.4 μmol/L)发挥抗病毒作用,提示其可能具有治疗方面的优势[36],2014年,Wilde等检测了氯喹对感染SARS-CoV的Vero E6细胞的抗病毒活性,EC50值为4.1μmol/L,CC50 >128 μmol/L,SI>31[37]。2017年,Kao等对50240种化合物进行筛选SARS-CoV小分子抑制剂,鉴定出喹啉类化合物32(图10),其在感染SARS-CoV的FRHK4细胞中测得的EC50值为8.4 μmol/L,在有效浓度下未观察到明显的细胞毒性[38]。
(6)吡啶类
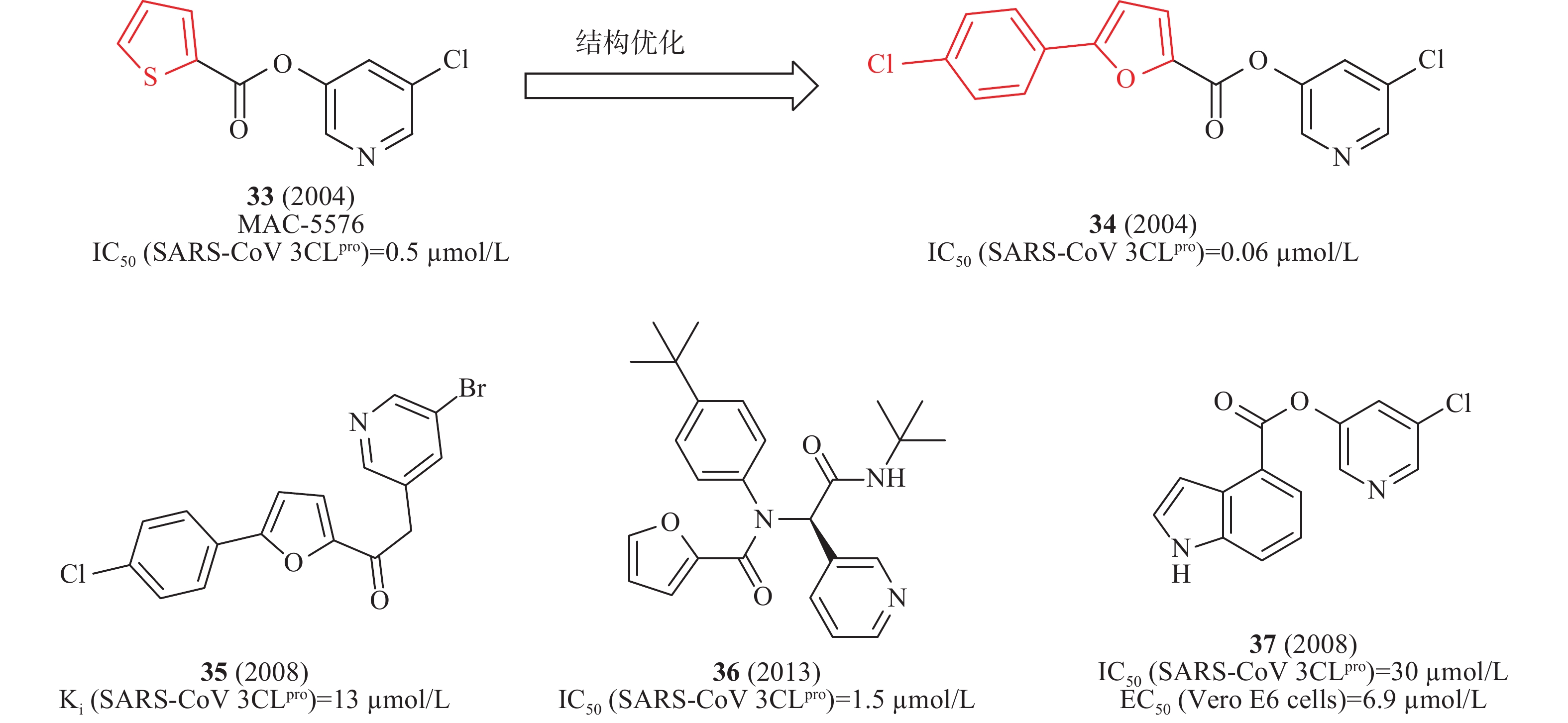
2008年,James等利用高通量筛选发现了化合物33(MAC-5576,图11)对SARS-CoV 3CLpro的酶活性IC50值为0.5 μmol/L;以33为先导化合物,基于分子对接技术设计合成了一系列衍生物,其中,含有氯苯取代的化合物34(图11)对SARS-CoV 3CLpro酶抑制活性提高了10倍(IC50值为0.06 μmol/L),表明SARS-CoV 3CLpro与呋喃一侧结合口袋具有较大疏水空腔,可以容纳苯环等大基团,为进一步结构优化提供了重要依据[39]。2008年,Vederas等制备一系列芳基亚甲基酮类似物,用FRET测试SARS 3CLpro抑制剂的酶活性。构效活性和35(图11)与SARS 3CLpro结合模型研究表明:3个芳香环对SARS-CoV 3CLpro具有良好抑制作用。该类化合物中35(图10)对IC50的抑制作用值为13 μmol/L[40]。2012年,Stauffer等利用NIH分子文库进行高通量筛选SARS-CoV 3CLpro抑制剂,得到探针化合物36(图11),在酶水平对SARS-CoV 3CLpro的IC50值为1.5 μmol/L,对SARS-CoV感染的Vero E6细胞抗病毒活性EC50值为12.9 μmol/L。此外,化合物36在中性PBS缓冲溶液溶解性较好(95 μg/ml)[41]。2008年,Mesecar等设计合成一系列5-氯吡啶酯衍生物,用FRET检测抗SARS-CoV 3CLpro的活性。构效关系表明,吲哚环第4位具有5-氯吡啶基酯的抑制剂37(图11)是最有效的抑制剂,在酶水平对SARS-CoV 3CLpro的IC50值为30 nmol/L,在感染SARS-CoV的Vero E6 细胞中测得EC50值为6.9 μmol/L[42]。
(7)其他类
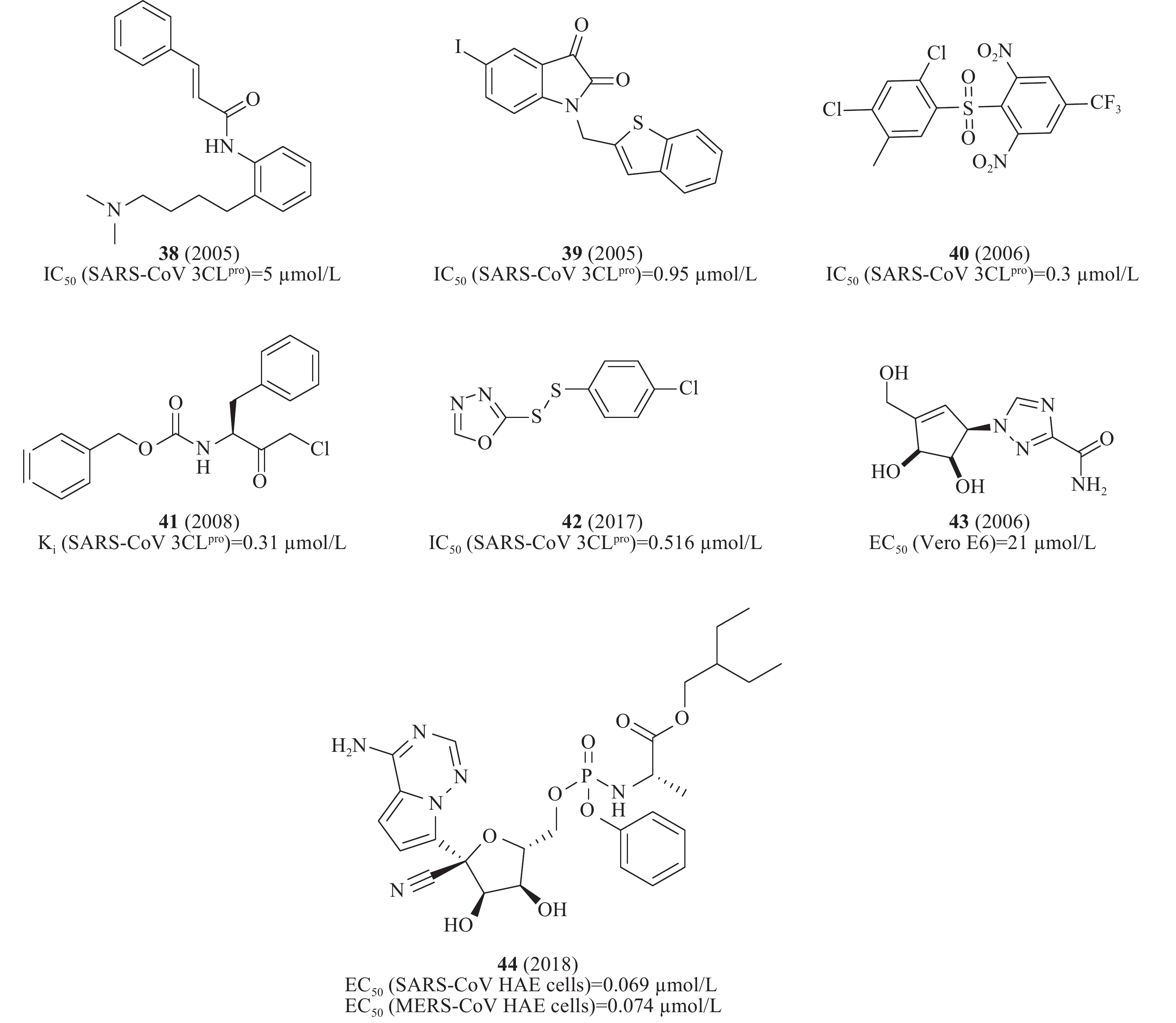
此外,通过虚拟筛选体外酶活性测试还发现一系列SARS-CoV 3CLpro小分子抑制剂,如血清素拮抗剂38(图12)(IC50= 5 μmol/L)[43]、异染色质衍生物39(图12)(IC50= 0.95 μmol/L)[44]、砜类化合物40(图12)(IC50= 0.3 μmol/L)[45]、卤甲基酮抑制剂41(图12)(Ki= 0.31 μmol/L)[46]、不对称的芳香族二硫醚化合物42(图12)(IC50= 0.516 μmol/L)[47]、非天然的五元环杂环碳环核苷1,2,4-三唑类似物43(图12)对SARS-CoV感染的Vero E6细胞EC50为21 μmol/L[48]。2018年,Denison等发现核苷类似物44(图12)(GS-5734,remdesivir)在体外和SARS-CoV小鼠模型中能有效抑制人和人畜共患的冠状病毒。在感染SARS-CoV的人气道上皮细胞(human airway epithelial cells, HAE)原代培养物中评估44的抗病毒活性,显示44的EC50值为0.069 μmol/L[49],通过定量逆转录聚合酶链反应发现,随着44剂量的增加,SARS-CoV感染的细胞内病毒基因组和亚基因组RNA数量均呈剂量依赖性减少,与病毒滴度降低一致[50]。
2.2 MERS-CoV的小分子抑制剂
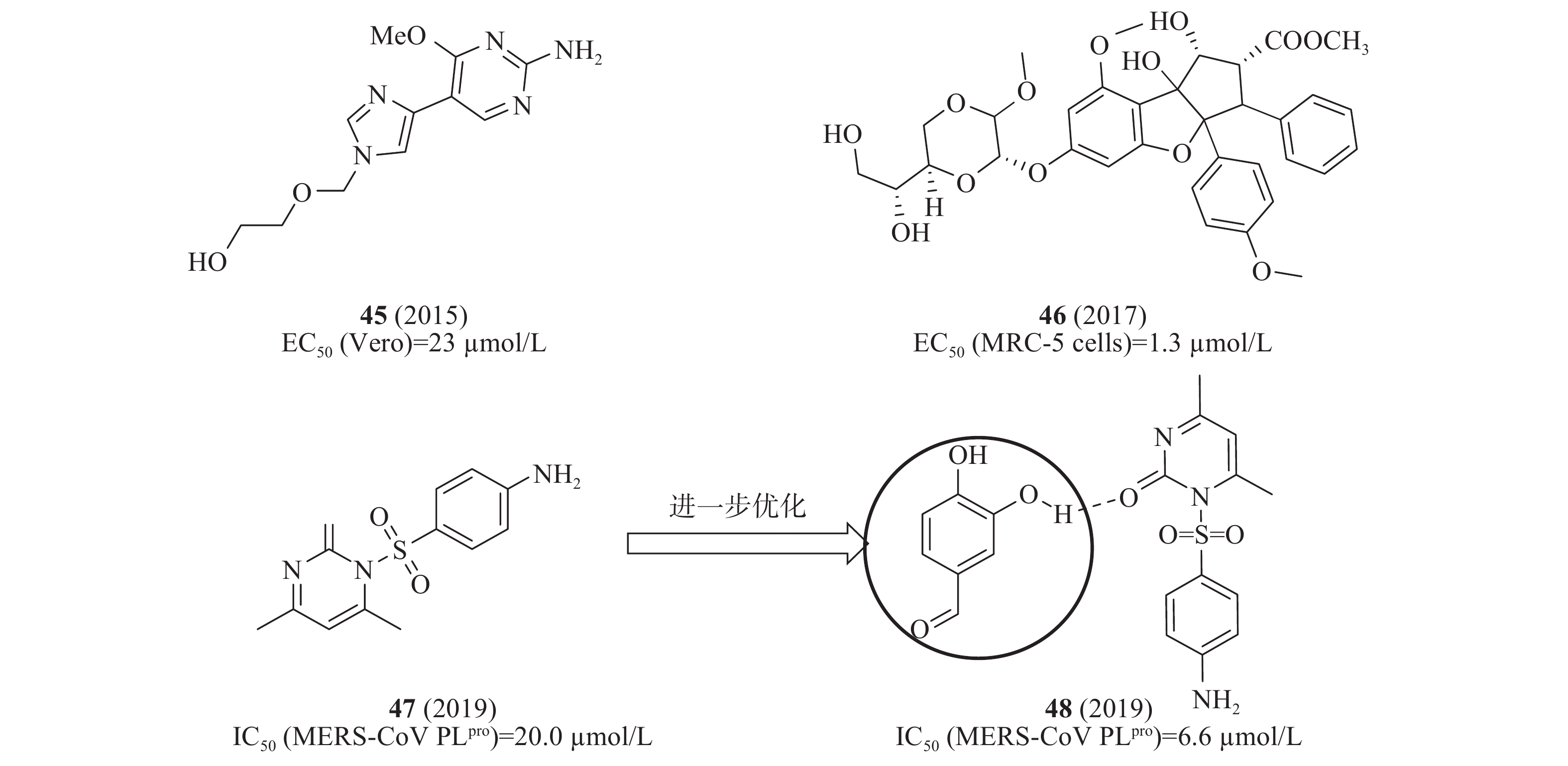
2015年,Radtke等设计一系列双柔性核苷类似物并测试其活性,其中,化合物45(图13)在感染MERS-CoV的Vero E6细胞中显示抗病毒活性EC50值为23 μmol/L[51]。2017年,Grünweller等用双重萤光素酶测定法和病毒感染的原代细胞分析化合物46(图13)对病毒翻译的影响,确定感染MERS-CoV的MRC-5细胞的EC50值为1.3 μmol/L;在MERS-CoV感染的人外周血单核原代细胞(PBMC)证实了化合物46的抗病毒活性[52]。2019年,Johnson等基于SPR和FRET技术,从含有30000种化合物的3种不同化合物库(Prestwick, Maybridge,Chembridge)中筛选出化合物47(图13),对MERS-CoV PLpro酶具有明显抑制活性,IC50值为20.0 μmol/L;为了进一步改善其抑制活性,从352个小片段库得到一个片段48(ZT626+47),48是由47和ZT626之间形成氢键作用结合在一起(图13),IC50值为6.6 μmol/L,比47提高了2倍,后续将通过分子对接研究47和ZT626的具体结合模式[53]。
前面提到的多个SARS-CoV 3CLpro对MERS-CoV也具有明显的抑制活性。例如,2014年De Wilde等对348个药物进行了抗MERS-CoV活性的筛选。研究表明化合物31(氯喹,图10)在体外感染MERS-CoV的Vero E6细胞中测得EC50值为3.0 μmol/L,CC50值为58.1 μmol/L,SI值为19.4[37]。2015年,Simmons等筛选含有2100个化合物的库,发现化合物11(图5)在酶水平上对MERS-CoV 3CLpro的活性最好,IC50值为46.12 μmol/L[20]。2016年,Liang等通过老药新用的方法筛选NA抑制剂对SARS-CoV的作用,发现该类化合物中24在酶水平(图8)对MERS-CoV 3CLpro IC50为12.2 μmol/L[31]。2017年,Kao等对50240种化合物进行筛选MERS-CoV小分子抑制剂,鉴定出化合物32(图10),其在感染MERS-CoV的FRHK4细胞中测得的EC50值为10.1 μmol/L[38]。2018年,Denison等发现核苷类似物44(图13)(GS-5734,remdesivir),研究人员选择在人气道上皮细胞(HAE)原代培养物中评估44对MERS-CoV的抗病毒活性,显示44对MERS-CoV的EC50值为0.074 μmol/L[49]。
2.3 SARS-CoV-2的小分子抑制剂
在当前SARS-CoV-2疫情下,运用“老药新用,一药多用”策略开展科技攻关研发可治疗COVID-19的药物,可大大缩短药物研发时间,进而可以解决新药研发周期长、难以满足当前疫情控制急需用药的难题。多个药物或候选药物(图14)被发现具有显著的抗SARS-CoV-2病毒效能,包括化合物49(利巴韦林,EC50= 109.50 μmol/L、CC50>400 μmol/L、SI>3.65),化合物50(喷昔洛韦,EC50= 95.96 μmol/L、CC50>400 μmol/L、SI> 4.17)、化合物51(硝唑尼特,EC50= 2.12 μmol/L、CC50> 35.53 μmol/L、SI> 16.76)、化合物52(萘莫司他,EC50= 22.50 μmol/L、CC50> 100 μmol/L、SI> 4.44)、化合物31(图10)(氯喹,EC50= 2.71 μmol/L、CC50> 211.70 μmol/L、SI> 100.81)、化合物44(图12)(瑞德西韦,GS-5734)、化合物53(法匹拉韦,T-705,EC50= 61.88 μmol/L、CC50> 400 μmol/L、SI> 6.46)、化合物54(羟氯喹,EC50= 4.51 μmol/L、CC50> 249.49 μmol/L、SI> 55.32)等[54-55]。
Gao等报道了一项多中心临床试验研究氯喹治疗100多例SARS-CoV-2感染的肺炎患者的效果。结果表明,化合物31在抑制肺炎恶化、改善肺部影像及缩短病程方面均优于对照组,且上述患者未见严重的不良反应,且毒性可以接受[56]。
Wang等在体外用非洲绿猴肾Vero E6细胞(ATCC-1586)测试羟氯喹的细胞毒性,结果显示,化合物54在体外可以有效抑制SARS-CoV-2感染,54是一种已知安全有效的抗炎药,预测该药物对COVID-19患者具有良好的抗炎潜力,这种可能性有待临床试验的证实[56]。Patel等2020年5月22日在《柳叶刀》上发表论文指出:COVID-19住院患者单独使用氯喹或羟氯喹或与大环内酯类抗生素联用在治疗新冠肺炎患者时,并未观察到益处;且有可能造成患者生存率降低和室性心律失常的频率增加[57],世卫组织随即宣布暂停相关临床试验,而该文数据的真实性被广泛质疑且未提供原始数据,该文章已被撤稿。世卫组织宣布重启相关临床实验。
化合物44作用于病毒进入细胞的后期阶段,与核苷类药物的作用机制一致。对感染Vero E6细胞的EC90= 1.76 μmol/L,具有较低不良反应,由同情用药过渡到随机双盲的严格III期临床试验(NCT04257656),治疗轻、中和重度感染COVID-19患者。报告显示,在针对237名病患的试验中,158人服用了瑞德西韦,另97人没有服用该药,结果服药患者的病死率为13.9%,没服药者的病死率为12.8%。此外,服用瑞德西韦的患者还出现了明显的副作用,病毒数量及症状却没有明显改善。王辰、曹彬团队得出的结论是:未观察到瑞德西韦联合标准疗法与标准疗法相比有统计学意义上显著的临床获益[58]。另外一项是由美国国立卫生研究院领衔的临床研究,从2020年2月21日开始,共招募1063名患者,随机分为试验组和安慰剂组,评价指标为康复时间。研究结果显示,接受瑞德西韦治疗的试验组患者痊愈中位时间为11 d、安慰剂组为15 d,与安慰剂组相比,接受瑞德西韦的试验组患者痊愈时间缩短31%,P<0.001,结果有显著统计学差异。但是,试验组患者病死率为8.0%,安慰剂组为11.6%,P= 0.059,病死率无显著的统计学差异[59]。
Zhou等发现,SARS-CoV-2与SARS-CoV基因组有79.5%的相似性[60]。基于二者序列同源性高,通过与相同受体ACE2(angiotensin converting enzyme 2)结合后进入细胞,且已有文献报道证实[61],55(洛匹那韦/利托那韦, lopinavir/ritonavir)在SARS的治疗中显示出良好的效果,提示可能对SARS-CoV-2的治疗有效。在重症COVID-19成人住院患者中,与常规治疗相比,未观察到化合物55对治疗有益 (临床试验注册号:ChiCTR2000029308)。试验结果表明,使用洛匹那韦/利托那韦组合与标准护理相比,并没有使严重COVID-19成人患者症状得到显著改善[62]。
化合物56(阿比多尔, arbidol)是一种具有免疫增强的非核苷类广谱抗病毒药物,用于治疗甲、乙型流感病毒等引起的上呼吸道感染。体外细胞实验显示:与药物未处理的对照组相比,阿比多尔在10~30 μmol/L浓度下,有效抑制SARS-CoV-2达60倍,并能显著抑制病毒对细胞的病变效应。目前已注册有盐酸阿比多尔片治疗COVID-19的临床研究(ChiCTR2000029621)。
3. 结语
到目前为止,冠状病毒没有特效的抗病毒药物或疫苗。新药研发通常需要大量的人力、物力、财力和时间;在当前COVID-19疫情急需药物的情况下,按照常规研发思路开展药物研发,显然“远水解不了近渴”。“老药新用”策略的优势就是可以节约资源和时间,化合物的安全性已经过验证,可直接进入临床药效研究。从战略药物储备角度长远考虑,我们还可以利用计算机辅助药物设计技术开展药物设计及化合物库筛选,发现新骨架、结构多样的苗头化合物,进而通过构效关系研究、结构优化及成药性评价,发现先导化合物、候选药物及临床有效药物,可丰富抗病毒药物的“储备库”。
-
图 2 雷公藤次碱对LPS诱导RAW264.7细胞分泌炎症因子的影响
A. NO光密度值;B. IL-1β光密度值;C. TNF-α光密度值;D. IL-6光密度值*P<0.05,**P<0.01,与模型组比较;##P<0.01,与空白组比较
图 3 雷公藤次碱对TRAF6表达和IRAK磷酸化的表达的影响
A. TRAF6磷酸化;B. IRAK磷酸化*P<0.05,**P<0.01,与模型组比较;##P<0.01,与空白组比较
图 4 雷公藤次碱对IκBα磷酸化和NF-κB核转位的影响
A. 细胞质NF-κB p65含量;B. 细胞核NF-κB p65含量;C. IκBα磷酸化*P<0.05,**P<0.01,与模型组比较;##P<0.01,与空白组比较
图 5 雷公藤次碱对p38, ERK和JNK MAPKs磷酸化的影响
A. JNK磷酸化;B. p38磷酸化;C. ERK磷酸化*P<0.05,**P<0.01,与模型组比较;##P<0.01,与空白组比较
-
[1] 白雪, 付瑞嘉, 乐世俊, 等. 雷公藤治疗类风湿性关节炎研究进展[J]. 中草药, 2020, 51(1):265-275. doi: 10.7501/j.issn.0253-2670.2020.01.034 [2] 姚骥如, 孙莹, 罗顺葵, 等. 雷公藤多苷的临床应用进展[J]. 中国新药与临床杂志, 2010, 29(3):179-182. [3] 胡攀勇, 李振麟, 濮社班, 等. 雷公藤研究进展[J]. 中国野生植物资源, 2013, 32(2):1-3,7. doi: 10.3969/j.issn.1006-9690.2013.02.001 [4] ZHANG Y Q, XU W, LI H, et al. Therapeutic effects of total alkaloids of Tripterygium wilfordii Hook f. on collagen-induced arthritis in rats[J]. J Ethnopharmacol,2013,145(3):699-705. doi: 10.1016/j.jep.2012.11.018 [5] 刘莉, 闫君, 舒积成, 等. 雷公藤生物碱类成分及其药理活性研究进展[J]. 天然产物研究与开发, 2019, 31(12):2170-2181. doi: 10.16333/j.1001-6880.2019.12.022 [6] LI T, LI F, LIU X Y, et al. Synergistic anti-inflammatory effects of quercetin and catechin via inhibiting activation of TLR4-MyD88-mediated NF-κB and MAPK signaling pathways[J]. Phytother Res,2019,33(3):756-767. doi: 10.1002/ptr.6268 [7] 鲍璐璐, 崔立红. TLR4/MyD88/NF-κB信号通路的研究进展[J]. 胃肠病学和肝病学杂志, 2019, 28(5):568-572. doi: 10.3969/j.issn.1006-5709.2019.05.019 [8] 师宝君, 姬志勤, 张继文, 等. 昆明山海棠的杀虫活性及有效成分[J]. 昆虫学报, 2007, 50(8):795-800. doi: 10.3321/j.issn:0454-6296.2007.08.007 [9] LAI J L, LIU Y H, LIU C, et al. Indirubin inhibits LPS-induced inflammation via TLR4 abrogation mediated by the NF-kB and MAPK signaling pathways[J]. Inflammation,2017,40(1):1-12. doi: 10.1007/s10753-016-0447-7 [10] LIU F, SUN G Q, GAO H Y, et al. Angelicin regulates LPS-induced inflammation via inhibiting MAPK/NF-κB pathways[J]. J Surg Res,2013,185(1):300-309. doi: 10.1016/j.jss.2013.05.083 [11] 单佳铃, 程虹毓, 文乐, 等. TLR/MyD 88/NF-κB信号通路参与不同疾病作用机制研究进展[J]. 中国药理学通报, 2019, 35(4):451-455. doi: 10.3969/j.issn.1001-1978.2019.04.002 [12] 崔嵩, 刘学锋, 吴斌. NF-кB在肿瘤中的研究进展[J]. 现代肿瘤医学, 2009, 17(1): 134-137. 范慧婕, 谭章斌, 赵晓山, 等. 四逆散通过MAPKs/NF-κB途径保护脂多糖致Raw264.7的细胞炎症[J]. 暨南大学学报(自然科学与医学版), 2019, 40(1): 10-18. [13] KAMINSKA B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy: from molecular mechanisms to therapeutic benefits[J]. Biochim Biophys Acta,2005,1754(1-2):253-262. doi: 10.1016/j.bbapap.2005.08.017 期刊类型引用(1)
1. 廖威权,李禹莹,李志岭,郭盈澳,谢静静,张剑勇. 基于网络药理学、SMR及分子对接探讨昆仙胶囊治疗强直性脊柱炎的作用机制. 中药新药与临床药理. 2025(03): 405-415 .  百度学术
百度学术其他类型引用(2)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 11319
- HTML全文浏览量: 3666
- PDF下载量: 135
- 被引次数: 3

