

 下载:
下载:
 下载:
下载:

-
肾衰宁颗粒由太子参、黄连、制半夏、陈皮、茯苓、大黄、丹参、牛膝、红花、甘草等十味中药制成;具有补气健脾,活血化痰,祛浊的功效[1]。有文献表明,肾衰宁在治疗慢性肾脏疾病中疗效较为显著[2];在尿毒症腹膜透析患者的治疗过程中能够降低血清硫酸吲哚酯浓度[3];对于慢性肾脏病的Ⅳ期患者,在西医基础治疗的同时服用肾衰宁颗粒,可以明显改善肾功能,同时提升患者的治愈率,具有较高的临床使用价值[4]。由于中药成分复杂[5],使得如何控制中药的质量成为十分重要的问题。指纹图谱是在了解中药物质整体作用的基础上,通过光谱和色谱技术获得中药化学成分的光谱或色谱图,以鉴别中药的真伪,评价质量的一致性和产品的稳定性,其具有信息量大、特征性强、完整性和模糊性等特点[6]。指纹图谱中的质量控制技术既能保证中药的功效,又在实现中药现代化过程中起关键性作用[7]。因此,本实验以10个批次的肾衰宁颗粒为研究对象,拟建立肾衰宁颗粒的指纹图谱,对肾衰宁颗粒进行质量评价。
肾衰宁颗粒是由十味中药组成的复方制剂,其化学成分十分复杂,且许多复方治疗疾病的药物基础并不明显,因此检测成分必须是发挥药效的有效成分[8],本实验针对大黄中的大黄酚(chrysophanol)[9-10]、丹参中的丹酚酸B(salvianolic acid B)[11]、陈皮中的橙皮苷(hesperidin)[9, 12]3种指标性成分,进行HPLC法含量测定。在多成分的质量控制检测成本高而对照品紧缺的情况下,能较大程度地节约检验成本,又可较全面地控制该制剂的质量,保证临床用药的有效性和安全性,同时为肾衰宁颗粒的质量控制提供参考。
-
Agilent 1260高效液相色谱仪(美国Agilent公司),包含G1311C四元泵,G1329B自动进样器,G1316A柱温箱,G4212B-DAD二极管阵列检测器,Chemstation色谱工作站;光电分析天平(德国Sartorius公司,CPA 225D型),最大载荷220 g,分度值0.01 mg;冷冻真空浓缩仪(丹麦Labogene公司,ScanVac ScanSpeed 40型);超声波清洗器(上海科导超声仪器有限公司,SK7200H型);涡旋混匀器(海门市其林贝尔仪器制造有限公司,Vortex QL-901型)。
-
肾衰宁颗粒(德元堂制药集团,批号:51103111、51103009 346、51103010 471、51103011 593、51103105、51103018 486、41103033 563、51103110、61103102、61103101)。蜕皮激素(ecdysterone,批号:P11N6F5706)、甘草苷(liquiritin,批号:2O1027BA14)、甘草酸(glycyrrhizic acid,批号:230A6B1)、大黄酚(chrysophanol,批号:T31O6F5345)、丹酚酸B(salvianolic acid B,批号:Y14M7H14804)、橙皮苷(hesperidin,批号:K02M3C1)对照品,均由上海源叶有限公司提供。大黄素(modin,批号:110756-200110)、盐酸小檗碱(berberine hydrochloride,批号:09030522)对照品,由中国食品药品检定研究院提供。甲醇、乙腈、甲酸,均为德国Merck公司生产,色谱纯。二氯甲烷,色谱纯。水为纯净水,娃哈哈公司生产。
-
色谱柱:Waters SunFire™ C18(250 mm×4.6 mm,5 μm)。流动相A:乙腈;流动相B:0.1%甲酸溶液,梯度洗脱。流速:1 ml/min。柱温:25 ℃。进样量:10 μl。检测波长:254 nm。梯度洗脱条件见表1。
表 1 梯度洗脱条件
时间(t/min) 乙腈(%) 0.1%甲酸溶液(%) 0 5 95 1 23 77 18 25 75 19 30 70 31 75 25 60 85 15 -
精密依次称取丹酚酸B、橙皮苷、大黄素标准品10.25、10.35、10.05 mg,分别置于10 ml容量瓶中,以纯甲醇定容至刻度,摇匀,得储备液并将其分装后储存于−20 ℃的冰箱中。精密称取10.10 mg大黄酚,置于10 ml容量瓶中,加入少量二氯甲烷溶解,超声处理3 min,然后用纯甲醇稀释至刻度,摇匀,得到对照品的储备液,置于−20 ℃冰箱保存。
-
取肾衰宁颗粒适量,研磨成细粉,混合均匀,精密称取0.10 g,加适量70%甲醇溶液使其溶解,超声45 min,再用70%甲醇溶液定容至2 ml,冷却,过0.45 μm微孔滤膜后,取续滤液进样分析。
-
取肾衰宁颗粒(批号:41103033 563),按照“2.3”项制备供试品溶液,在“2.1”项色谱条件下连续进样5次,通过大黄素(7号色谱峰)作为参照峰,确定相对保留时间和相对峰面积。相对保留时间的RSD在1.0%以内,相对峰面积的RSD在2.0%以内,表明进样仪器的精密度良好。
-
取肾衰宁颗粒(批号:41103033 563),按照“2.3”项平行制备5份供试品溶液,测定在“2.1”项色谱条件下的相对保留时间和相对峰面积。相对保留时间RSD在1.0%以内,相对保留面积RSD在2.0%以内。表明该实验方法的重复性良好。
-
取肾衰宁颗粒(批号:41103033 563),并根据“2.3”项下平行制备5份供试品溶液,在“2.1”项色谱条件下,分别记录0、4、8、24、36 h的相对保留时间和相对峰面积。相对保留时间RSD在1.0%以内,相对保留面积RSD在2.0%以内。表明供试样品溶液在36 h内稳定性好。
-
分别称取10个批次的肾衰宁粉末,按照“2.3”项制备供试品溶液,每个批次平行制备3份供试品溶液,记录图谱,见图1。利用中国药典委员会“中药色谱指纹图谱相似度评价系统2008A版”的软件,将10批次肾衰宁颗粒的图谱导入,将批号为61103102的样品溶液用作针对相似性计算校正的参考图。结果显示,1~9批制剂间指纹图谱与对照图谱之间相似度均不小于0.90,见表2,表明相似度良好。对保留时间0~60 min内的色谱峰进行分析,均有22个稳定的特征峰,确定其为肾衰宁的共有指纹峰,经过标准品比对,其中,6号峰为橙皮苷,14号峰为丹酚酸B,21号峰为大黄酚。

图 1 10批肾衰宁颗粒的色谱图
表 2 10批肾衰宁颗粒HPLC指纹图谱相似度
样品号 S1 S2 S3 S4 S5 S6 S7 S8 S9 S10 对照指纹图谱 S1 1.000 0.980 0.910 0.941 0.934 0.948 0.916 0.941 0.950 0.874 0.965 S2 0.980 1.000 0.934 0.934 0.932 0.945 0.951 0.946 0.978 0.867 0.973 S3 0.910 0.934 1.000 0.952 0.947 0.937 0.972 0.940 0.945 0.914 0.973 S4 0.941 0.934 0.952 1.000 0.990 0.984 0.951 0.971 0.923 0.940 0.986 S5 0.934 0.932 0.947 0.990 1.000 0.978 0.938 0.976 0.910 0.910 0.979 S6 0.948 0.945 0.937 0.984 0.978 1.000 0.954 0.987 0.931 0.923 0.986 S7 0.916 0.951 0.972 0.951 0.938 0.954 1.000 0.949 0.966 0.913 0.979 S8 0.941 0.946 0.940 0.971 0.976 0.987 0.949 1.000 0.928 0.890 0.980 S9 0.950 0.978 0.945 0.923 0.910 0.931 0.966 0.928 1.000 0.892 0.969 S10 0.874 0.867 0.914 0.940 0.910 0.923 0.913 0.890 0.892 1.000 0.937 对照指纹图谱 0.965 0.973 0.973 0.986 0.979 0.986 0.979 0.980 0.969 0.937 1.000 -
按照“2.2”项下制备标准品溶液,得到混合对照品色谱图见图2。将标准品储备液用纯甲醇稀释,丹酚酸B浓度梯度为:10、20、40、80和100 μg/ml;橙皮苷浓度梯度为:40、80、160、320和400 μg/ml;大黄酚浓度梯度为:8、16、40、100和350 μg/ml。在“2.1”项色谱条件下,分别记录3种成分的峰面积,以峰面积(Y)对浓度(X)进行线性回归,结果见表3。
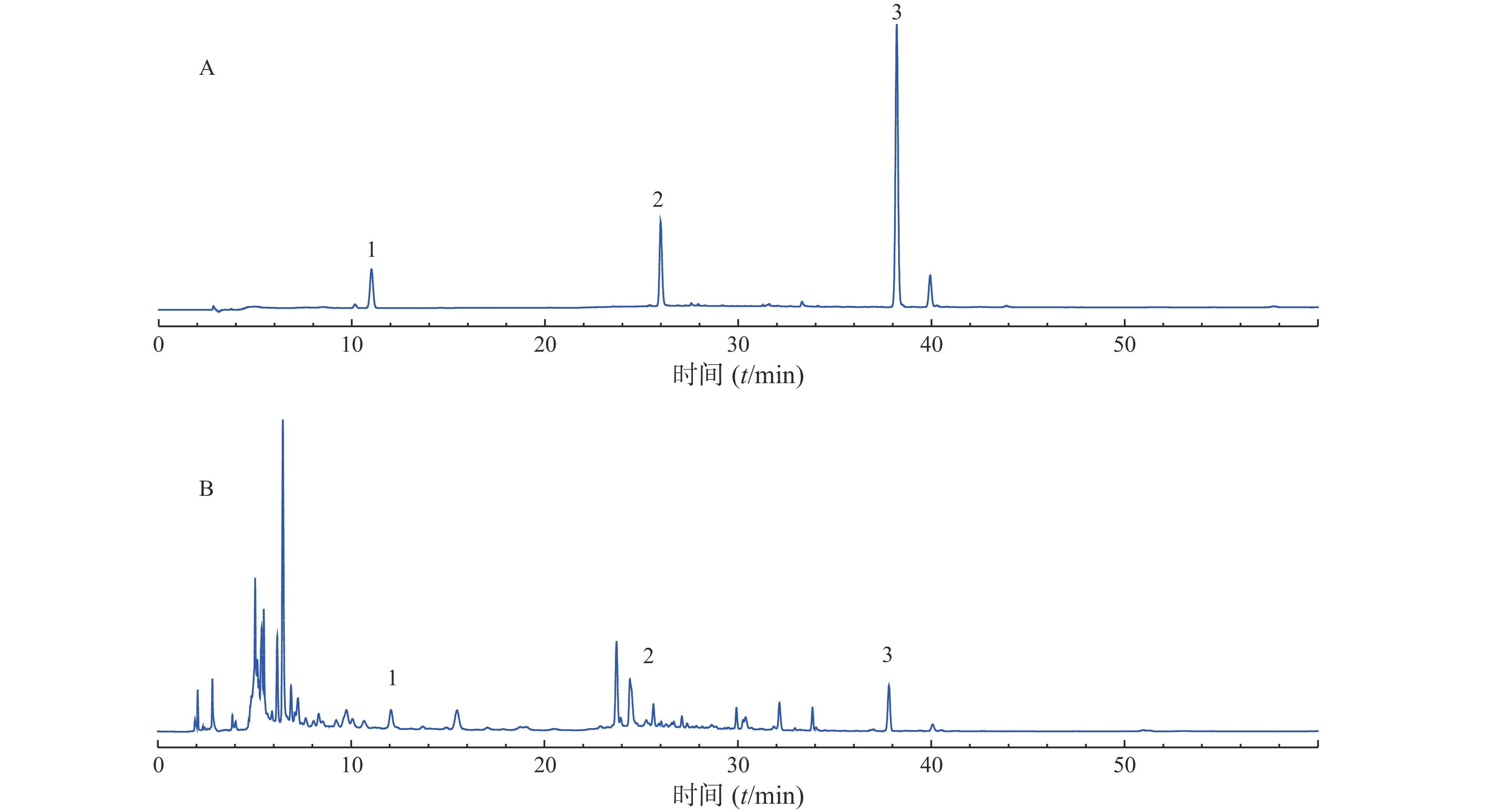
图 2 混合对照品溶液(A)和供试品溶液(B)色谱图
表 3 各成分线性关系
对照品 回归方程 r 线性范围(μg/ml) 橙皮苷 Y=2.391 8X–2.798 3 0.999 7 40~400 丹酚酸B Y=10.689X–5.578 3 0.999 8 10~100 大黄酚 Y=58.983X+121.99 0.999 9 7~350 -
取同一对照品溶液,根据“2.1”项下色谱条件进行测定,重复进样5次,并记录峰面积。橙皮苷、丹酚酸B、大黄酚峰面积的RSD分别为0.17%、0.20%、0.15%,表明仪器精密度良好。
-
精密吸取橙皮苷、丹酚酸B、大黄酚对照品溶液适量,使用甲醇逐步稀释,至色谱图中上述3种成分的峰高分别为噪音的3倍和10倍,测得橙皮苷的检测限和定量限分别为0.18和1 μg/ml;丹酚酸B的检测限和定量限分别为0.3和1.2 μg/ml;大黄酚的检测限和定量限分别为1和5 μg/ml。
-
取同一供试品溶液(批号:41103033 563),并根据“2.1”项色谱条件在0、2、4、8、24、36 h测定。橙皮苷、丹酚酸B、大黄酚的峰面积的RSD分别为2.01%、2.22%、2.05%,结果表明:供试品在36 h内稳定。
-
取同一供试品溶液(批号:41103033 563),平行制备6份,并根据“2.1”项色谱条件进行测定,橙皮苷、丹酚酸B、大黄酚的峰面积的RSD分别为0.67%、2.00%、2.02%,表明系统重复性良好。
-
精密量取肾衰宁溶液1 ml,分别加入100%含量橙皮苷对照品(理论值为192.02 μg),100%含量丹酚酸B对照品(理论值为62.66 μg),100%含量大黄酚对照品(理论值35.22 μg),按照“2.2”项平行制备6份,并根据“2.1”的色谱条件进行测量,按照下述公式计算加样回收率,平均加样回收分别为119.20%、84.69%、84.58%。结果见表4。
表 4 加样回收率试验结果
成分 取样量(V/ml) 样品含量(m/μg) 加入量(m/μg) 测得量(m/μg) 回收率(%) 平均回收率(%) 橙皮苷 1.00 383.02 95.88 773.46 101.80 100.27 1.00 380.39 95.88 764.91 100.26 1.00 380.31 95.88 763.64 99.95 1.00 384.46 95.88 771.17 100.83 1.00 385.81 95.88 772.95 100.94 1.00 389.20 95.88 764.32 97.81 丹酚酸B 1.00 120.79 31.80 250.42 101.91 98.82 1.00 122.50 31.80 251.14 101.12 1.00 125.56 31.80 248.78 96.87 1.00 127.57 31.80 254.87 100.07 1.00 127.74 31.80 251.44 97.25 1.00 127.75 31.80 249.44 95.66 大黄酚 1.00 72.67 17.79 138.19 92.06 97.36 1.00 69.62 17.79 135.41 92.44 1.00 68.46 17.79 140.83 101.69 1.00 71.03 17.79 141.25 98.66 1.00 68.98 17.79 141.57 102.00 1.00 71.92 17.79 141.17 97.30 回收率=(实测量-样品含量)/加入量×100%。
-
取10批肾衰宁颗粒,按“2.2”项制备试液,按“2.1”项色谱条件测定,结果见表5。
表 5 10批肾衰宁样品中3种成分的测定结果
药品批号 橙皮苷(μg/ml) 丹酚酸B(μg/ml) 大黄酚(μg/ml) 61103102 66.99 42.29 44.65 51103110 83.41 39.34 42.89 51103111 94.75 41.36 44.51 51103009 346 80.57 43.21 43.44 51103011 593 83.51 42.04 42.58 51103018 486 79.94 41.94 42.96 51103010 471 90.03 41.88 44.03 41103033 563 83.48 43.01 43.94 61103101 85,92 42.93 43.85 51103105 88.04 41.76 42.94 -
在190~400 nm处全波长扫描,盐酸小檗碱、大黄素、大黄酚、丹酚酸B、蜕皮激素、甘草酸、甘草苷、橙皮苷分别在345、254、254、286、250、237、237、283 nm处有最大吸收,通过对比分析,在波长254 nm处,上述8种成分具有良好的响应,样品中相邻色谱峰的基线分离可以满足含量检测的要求。因此,选择波长254 nm作为检测波长。
-
将肾衰宁粉末直接用水溶解后进样,发现样品出峰很少,所测成分在色谱条件下没有吸收。后用50%、70%、80%的甲醇溶解样品,发现用70%甲醇溶解出峰数最多,且所测成分在此条件下均有吸收,故选择70%甲醇进行样品的前处理。
-
有文献报道,肾衰宁含量测定使用甲醇-0.1%磷酸[13]、乙腈-水-1%甲酸[14]、乙腈-05%磷酸[15]进行90 min的大梯度洗脱,发现色谱峰分不开,且特征峰较少。经过反复实验,确定乙腈-0.1%甲酸水溶液用作梯度洗脱的流动相,分离出样品中测得的特征峰,特征峰很多,峰形良好。
Research on the fingerprint and three active components assay in Shenshuaining granules by HPLC
-
摘要:
目的 通过高效液相色谱法建立肾衰宁颗粒的指纹图谱,并测定肾衰宁颗粒中3种有效成分(橙皮苷、丹酚酸B和大黄酚)的含量,为肾衰宁颗粒的质量控制提供依据。 方法 采用SunFireTM C18色谱柱,乙腈-0.1%甲酸水溶液为流动相,梯度洗脱,流速1 ml/min,检测波长254 nm,柱温25 ℃,建立肾衰宁颗粒的指纹图谱,并对3种有效成分的含量进行测定。 结果 测定10批肾衰宁颗粒的色谱图,确立的标准指纹图谱上有22个共有峰,各批次的指纹图谱与对照图谱之间相似度均不小于0.90,相似度良好。方法学验证结果表明重复性、稳定性、精密度结果均符合要求。 结论 本法操作简单、可靠,适用于肾衰宁颗粒指纹图谱的建立及含量测定,为肾衰宁颗粒的质量控制提供依据。 Abstract:Objective To establish the fingerprint spectrum and assay three active components (hesperidin, salvianolic acid B and chrysophanol) in Shenshuaining granule by HPLC method. Methods The chromatographic separation was achieved on SunFireTM C18 column with acetonitrile-0.1% formic acid aqueous solution as mobile phase. Gradient elution program was applied with flow rate of 1.0 ml/min, detection wavelength at 254 nm and the column temperature at 25 ℃. The fingerprint spectrum was established and three active components in Shenshuaining granule were assayed. Results There were 22 common peaks on the fingerprints after analyzing chromatograms from 10 batches of Shenshuaining granules. Good fingerprint similarities (≥0.9) between different batches and the control chromatogram were found. This method has great repeatability, stability and precision, which meets all the assay requirements. Conclusion A simple and reliable HPLC method was developed, which is suitable for the fingerprint establishment of Shenshuaining granules. It provides a method for the quality control of Shenshuaining granules. -
Key words:
- Shenshuaining granules /
- HPLC /
- fingerprinting /
- content determination
-
放线菌以其能产生结构新颖且有良好生物活性的先导化合物而备受关注[1],一直被认为是天然药物的重要生产者,其主要结构类型包括聚酮、生物碱、多肽和萜烯类化合物等,同时涵盖了多种多样的生物活性如抗菌、抗寄生虫、免疫调节、抗炎、抗癌等[2-4],这突显了放线菌具有不可预估的药物开发潜力。
随着研究的深入,陆地和普通环境中的资源日趋枯竭,很多微生物及其次生代谢产物被重复开发和提取分离,发现新活性分子的几率愈来愈低,开发创新药物的难度越来越大[5-6];而极地极端的生态环境造就的微生物具有产生更为特别的化学骨架和活性次生代谢产物的能力,是新型药源分子的重要来源。本文以采自北极楚克奇海域海绵共附生放线菌Streptomyces sp. LHW11-07为研究对象,从其发酵浸膏中分离鉴定了9个单体化合物1~9(图1),其中化合物1和2为该属内首次分离得到。
1. 材料和方法
1.1 实验仪器与试剂
AMX-600型核磁共振仪(德国Bruker公司);Xevo G2-XS Q-TOF液质联用仪、1525/2996, 2998型高效液相色谱仪(美国Waters公司);半制备型HPLC色谱柱(Atlantis Prep T3,美国Waters公司;YMC C18,日本YMC公司);中压柱色谱仪(法国Interchim公司);恒温振荡培养箱(上海一恒科学仪器有限公司);N-1000型旋转蒸发仪(上海爱郎仪器有限公司);反相ODS硅胶和Sephadex LH-20柱色谱填料(Pharmacia公司);正相硅胶(200-300目)和TLC薄层板(烟台江友硅胶开发有限公司);分析级试剂(上海化学试剂公司);色谱级试剂(德国Merck公司);氘代试剂(美国剑桥同位素实验室公司)。
1.2 菌株的来源及鉴定
菌株分离于北极海域来源的海绵样本,经16S rRNA基因序列鉴定为Streptomyces sp.,编号为LHW11-07,菌种保存于上海交通大学医学院附属仁济医院药学部海洋药物研究中心。
1.3 菌株的大发酵
培养基为ISP2:葡萄糖(4 g/L)、酵母提取物(4 g/L)、麦芽糖提取物(10 g/L)以及海盐(25 g/L),加水溶解后调节pH为7.2~7.4,分装后高压灭菌20 min (121 ℃),冷却备用。
挑取Streptomyces sp. LHW11-07单菌落至1级种子培养基里(100 ml ISP2培养基至250 ml三角瓶),置于30 ℃,220 r/min的恒温摇床培养3 d,得1级种子液;将1级种子液按5%接种量接到2级种子培养基里(150 ml ISP2培养基至500 ml三角瓶),置于30 ℃,220 r/min的恒温摇床培养3 d,得2级种子液;将2级种子液按5%接种量接到大发酵培养基里(700 ml ISP2培养基至2 L三角瓶),置于30 ℃,220 r/min的恒温摇床培养7 d,共得到发酵液50 L。
1.4 发酵产物的提取与分离
菌株培养7 d后,用等体积的乙酸乙酯萃取3次,合并乙酸乙酯提取液,减压浓缩得粗浸膏9.8 g。粗浸膏先经凝胶柱分离,二氯甲烷:甲醇(1∶1)的混合溶剂进行洗脱,得到组份Fr.1~Fr.7。
组份Fr.4经正相中压柱色谱分离(二氯甲烷:甲醇100:0~0:100),得到组份Fr.4a~Fr.4j。组份Fr.4d再经反相中压柱色谱分离(10%~100%乙腈水),得到组份Fr.4d1~Fr.4d8,Fr.4d2和Fr.4d3用反相半制备HPLC纯化(35%甲醇水,YMC C18),分别得到化合物1 (12 mg, tR=26 min)和2 (47.6 mg, tR=30 min);Fr.4d6用反相半制备HPLC纯化(49%甲醇水,YMC C18),得到化合物3 (8.4 mg, tR=23 min)和4 (2.0 mg, tR=30 min);组份Fr.4g再经凝胶柱纯化,洗脱剂为正己烷:二氯甲烷:甲醇(4∶5∶1),得到组份Fr.4g1~Fr.4g11,其中Fr.4g3用反相半制备HPLC纯化(25%乙腈水,YMC C18),得到化合物5 (1.2 mg, tR=18 min)。
组份Fr.7经反相中压柱色谱分离(10%~100%乙腈水),得到组份Fr.7A~Fr.7D。组份Fr.7A用反相半制备HPLC纯化(20%乙腈水,YMC C18),得到化合物6 (18 mg, tR=19 min)和7 (3.4 mg, tR=25 min);组份Fr.7B经正相中压柱分离(石油醚:丙酮100:0~0:100),得到组份Fr.7B1~Fr.7B9,Fr.7B4用反相半制备HPLC纯化(88%乙腈水,Atlantis Prep T3),得到化合物8 (12 mg, tR=23 min)和9 (8.4 mg, tR=26 min)。
2. 结构鉴定
化合物1:淡黄色固体,ESI-MS显示准分子离子峰m/z 372 [M+Na]+。1H-NMR (600 MHz, DMSO)显示在低场区有吲哚环的特征信号δH 7.00 (1H, s, H-2),7.49 (1H, d, J=8.0 Hz, H-4),7.02 (1H, t, J=7.6 Hz, H-5),7.05 (1H, t, J=7.6 Hz, H-6),7.32 (1H, d, J=8.0 Hz, H-7);4个活泼氢质子信号δH 10.89 (1H, d, J=2.6 Hz, NH-1),7.83 (1H, d, J=3.0 Hz, NH-10),7.62 (1H, d, J=3.0 Hz, NH-13),9.20 (1H, s, OH-19);4个芳香质子信号δH 6.53 (2H, d, J=8.4 Hz, H-17, H-21),6.59 (2H, d, J=8.4 Hz, H-18, H-20),提示分子中有1个对位二取代的苯环;在高场区有两组亚甲基质子信号δH 2.80 (1H, dd, J=14.5, 4.5 Hz, H-8),2.43 (1H, ov, H-8),δH 1.83 (1H, dd, J=13.4, 6.9 Hz, H-15),2.47 (1H, ov, H-15),两个次甲基质子信号δH 4.01 (1H, m, H-9),3.95 (1H, m, H-12)。13C-NMR (150 MHz, DMSO)结合DEPT谱表明其有20个碳信号,2个酮羰基碳δC 166.7,166.2,14个芳香碳,2个亚甲基碳δC 30.0,40.0和2个次甲基碳δC 55.9,55.2。对其碳信号进行归属:δC 118.7 (C-2)、108.9 (C-3)、127.5 (C-3a)、118.4 (C-4)、120.8 (C-5)、124.3 (C-6)、111.3 (C-7)、136.0 (C-7a)、30.0 (C-8)、55.9 (C-9)、166.7 (C-11)、55.2 (C-12)、166.2 (C-14)、40.1 (C-15)、126.4 (C-16)、130.7 (C-17, C-21)、114.9 (C-18, C-20)、156.0 (C-19)。以上数据与文献[7]对比基本一致,故确定为cyclo-(L-Tyr-L-Trp)。
化合物2:淡黄色固体,ESI-MS显示准分子离子峰m/z 274 [M+H]+。1H-NMR (600 MHz, DMSO)发现其与化合物1一样有吲哚环的特征信号δH 7.09 (1H, s, H-2),7.52 (1H, d, J = 7.9 Hz, H-4),6.99 (1H, t, J = 7.8 Hz, H-5),7.02 (1H, t, J = 7.8 Hz, H-6),7.30 (1H, d, J = 7.8 Hz, H-7);3个活泼氨基质子信号δH 10.87 (1H, s, NH-1),δH 8.30 (1H, m, NH-10),7.85 (1H, d, J = 2.9 Hz, NH-13);两组亚甲基质子信号δH 3.21 (1H, m, H-8),3.13 (1H, m, H-8),δH 3.65 (1H, m, H-15),3.05 (1H, m, H-15),两个次甲基质子信号δH 4.87 (1H, m, H-9),4.00 (1H, m, H-12)。13C-NMR (150 MHz, DMSO)结合DEPT谱表明其有14个碳信号,2个酮羰基碳δC 167.2,165.7,8个芳香碳,2个亚甲基碳δC 63.0,30.3和2个次甲基碳δC 57.3,55.5。对其碳信号进行归属:δC 127.6 (C-2)、111.2 (C-3)、136.0 (C-3a)、118.6 (C-4)、120.8 (C-5)、124.0 (C-6)、118.3 (C-7)、109.0 (C-7a)、30.3 (C-8)、57.3 (C-9)、167.2 (C-11)、55.5 (C-12)、165.7 (C-14)、63.0 (C-15)。以上数据与文献[8]对比基本一致,故确定为cyclo-(L-Trp-L-Ser)。
化合物3:白色固体,ESI-MS显示准分子离子峰m/z 261 [M+H]+。1H-NMR (600 MHz, DMSO)提示有2个活泼氢质子信号δH 7.87 (1H, s, NH-8),9.22 (1H, s, OH-4’);1组对位二取代的苯环芳香质子信号δH 7.04 (2H, d, J = 8.2 Hz, H-2’, H-6’),6.63 (2H, d, J = 8.2 Hz, H-3’, H-5’);2个次甲基质子信号δH 4.24 (1H, t, J = 8.2 Hz, H-6),4.03 (1H, dd, J = 9.9, 2.9 Hz, H-9);4组亚甲基质子信号δH 3.42 (1H, m, H-3),3.24 (1H, m, H-3),1.73 (2H, m, H2-4),2.00 (1H, m, H-5),1.41 (1H, m, H-5),2.93 (2H, m, H2-10)。13C-NMR (150 MHz, DMSO)结合DEPT谱表明其有14个碳信号,2个酮羰基碳δC 168.9,165.1,6个芳香碳,2个次甲基碳δC 58.4,56.0以及4个亚甲基碳δC 44.6,34.7,27.8,21.9。对其碳信号进行归属:δC 165.1 (C-1)、44.6 (C-3)、21.9 (C-4)、27.8 (C-5)、58.4 (C-6)、168.9 (C-7)、56.0 (C-9)、34.7 (C-10)、127.0 (C-1’)、130.8 (C-2’, C-6’)、114.8 (C-3’, C-5’)、155.9 (C-4’)。以上数据与文献[9]对比基本一致,故确定为cyclo-(D-Tyr-D-Pro)。
化合物4:白色固体,ESI-MS显示准分子离子峰m/z 311 [M+H]+。1H-NMR (600 MHz, DMSO)显示3个活泼氢质子信号δH 9.30 (1H, s, OH-11),7.84 (2H, t, J = 2.9 Hz, NH-1, NH-4);9个芳香区质子信号:4个归为1组对位二取代苯环δH 6.85 (2H, d, J = 8.5 Hz, H-9, H-13),6.65 (2H, t, J = 8.5 Hz, H-10, H-12),5个归为1组单取代苯环δH 7.20 (1H, t, J = 7.6 Hz, H-18),7.04 (2H, d, J = 6.9 Hz, H-16, H-20),7.28 (2H, t, J = 7.6 Hz, H-17, H-19);2组亚甲基质子信号δH 2.58 (1H, dd, J = 13.6, 5.0 Hz, H-7),2.20 (1H, d, J = 6.5 Hz, H-7),2.19 (2H, dd, J = 13.6, 6.5 Hz, H2-14),2个次甲基质子信号δH 3.95 (1H, m, H-3),3.90 (1H, m, H-6)。13C-NMR (150 MHz, DMSO)结合DEPT谱显示其有18个碳信号,2个酮羰基碳δC 166.2,166.2,12个芳香碳,2个亚甲基碳δC 40.1,38.5和2个次甲基碳δC 55.7,55.4。对其碳信号进行归属:δC 166.2 (C-2)、55.7 (C-3)、166.3 (C-5)、55.4 (C-6)、40.1 (C-7)、126.5 (C-8)、130.8 (C-9, C-13)、115.0 (C-10, C-12)、156.1 (C-11)、38.5 (C-14)、136.7 (C-15)、129.7 (C-16, C-20)、128.2 (C-17, C-19)、126.4 (C-18)。以上数据与文献[10]对比基本一致,故确定为cyclo-(L-Tyr-L-Phe)。
化合物5:白色固体,ESI-MS显示准分子离子峰m/z 277 [M+H]+。1H-NMR (600 MHz, DMSO)显示3个活泼氢质子信号δH 9.22 (1H, s, OH-4’),8.02 (2H, dd, J = 5.6, 2.5 Hz, NH-1, NH-4);4个芳香质子信号δH 6.90 (2H, d, J = 8.2 Hz, H-2’, H-6’),6.64 (2H, d, J = 8.2 Hz, H-3’, H-5’),提示分子中有1个对位二取代苯环;3个次甲基质子信号δH 4.06 (1H, q, J = 3.3 Hz, H-3),3.44 (1H, m, H-6),1.43 (1H, ov, H-8),2个亚甲基质子信号δH 1.43 (1H, m, H-7),1.23 (1H, m, H-7),2.69 (1H, q, J = 13.6, 4.8 Hz, H-11),3.01 (1H, q, J = 13.7, 3.7 Hz, H-11)以及2个末端甲基质子信号δH 0.63 (6H, ov, H3-9, H3-10)。13C-NMR (150 MHz, DMSO)结合DEPT谱显示其有15个碳信号,2个酮羰基碳δC 166.2,167.4,6个芳香碳,2个亚甲基碳δC 43.7,37.7,3个次甲基碳δC 55.7,52.3,21.4以及2个甲基碳δC 22.9,22.8。对其碳信号进行归属:δC 166.2 (C-2)、55.7 (C-3)、167.4 (C-5)、52.3 (C-6)、43.7 (C-7)、21.4 (C-8)、22.9 (C-9)、22.8 (C-10)、37.7 (C-11)、125.8 (C-1’)、131.2 (C-2’, C-6’)、114.8 (C-3’, C-5’)、156.4 (C-4’)。以上数据与文献[11]对比基本一致,故确定为cyclo-(L-Tyr-L-Leu)。
化合物6:白色固体,ESI-MS显示准分子离子峰m/z 259 [M+Na]+。1H-NMR (600 MHz, DMSO)显示有2个活泼氢质子信号δH 5.05 (1H, d, J = 4.5 Hz, OH-4)和4.39 (1H, q, J = 4.0 Hz, OH-13),3个甲基质子信号δH 0.88 (3H, d, J = 6.8 Hz, H3-12),0.98 (3H, s, H3-14)和δH 1.05 (3H, s, H3-15),5对亚甲基质子信号δH 2.14 (1H, m, H-3),1.23 (1H, m, H-3),1.74 (1H, m, H-9),1.60 (1H, m, H-9),1.44 (3H, m, H2-10, H-11),1.32 (1H, d, J = 10.4 Hz, H-11),3.95 (2H, m, H2-13),3个次甲基质子信号δH 1.68 (1H, m, H-2),4.57 (1H, m, H-4),1.77(1H, m, H-8)。13C-NMR (150 MHz, DMSO)结合DEPT谱共显示有15个碳信号,包括4个季碳δC 52.1,150.1,136.7,39.8;3个次甲基碳δC 35.2,69.8,46.4;5个亚甲基碳δC 42.4,23.8,28.7,36.4,57.1以及3个甲基碳δC 13.7,29.1,24.4。对其碳信号进行归属:δC 52.1 (C-1)、35.2 (C-2)、42.4 (C-3)、68.9 (C-4)、150.1 (C-5)、136.7 (C-6)、39.8 (C-7)、46.4 (C-8)、23.8 (C-9)、28.7 (C-10)、36.4 (C-11)、13.7 (C-12)、57.1 (C-13)、29.1 (C-14)、24.4 (C-15)。以上数据与文献[12]对比基本一致,故确定为albaflavenol B。
化合物7:白色结晶固体,ESI-MS显示准分子离子峰m/z 268 [M+H]+。1H-NMR (600 MHz, DMSO)可看出其有13个氢信号,包括5个活泼氢质子信号δH 3.56 (2H, m, NH2),5.49 (1H, s, OH-5’),5.36 (1H, t, J = 4.9 Hz, OH-2’)和5.23 (1H, s, OH-3’),4个连氧次甲基质子信号δH 4.56 (1H, s, H-2’),4.14 (1H, s, H-3’),3.96 (1H, m, H-4’)和5.90 (1H, d, J = 5.8 Hz, H-1’),1组亚甲基信号δH 3.66 (2H, m, H2-5’)以及2个低场区的烯氢质子信号δH 8.37 (1H, s, H-8)和8.21 (1H, s, H-2)。13C-NMR (150 MHz, DMSO)显示其共有10个碳信号,结合DEPT谱可推测有3个芳香季碳δC 149.9,119.8,154.3,2个连氮的芳香次甲基碳δC 151.7,138.6,4个次甲基碳δC 87.8,73.5,70.5,85.7,1个亚甲基碳δC 61.5。对其碳信号进行归属:δC 151.7 (C-2)、149.9 (C-4)、119.8 (C-5)、154.3 (C-6)、138.6 (C-8)、87.8 (C-1’)、73.5 (C-2’)、70.5 (C-3’)、85.7 (C-4’)、61.5 (C-5’)。以上数据与文献[13]对比基本吻合,故确定为β-adenosine。
化合物8:绿色无定型固体,ESI-MS显示准分子离子峰m/z 297 [M+H]+。1H-NMR (600 MHz, MeOD)显示有12个氢信号,包括1个活泼氢质子信号δH 8.37 (1H, s, H-11),1个甲氧基质子信号δH 3.79 (3H, s),1个甲基质子信号δH 1.25 (3H, d, J = 6.4 Hz, H3-13),3个低场区的芳香氢质子信号δH 8.31 (1H, d, J = 7.8 Hz, H-4),6.92 (1H, t, J = 8.0 Hz, H-5)和7.65 (1H, d, J = 8.0 Hz, H-6),以及2个次甲基质子信号δH 4.74 (1H, d, J = 3.2 Hz, H-8)和4.40 (1H, m, H-12),后与文献[14]对比发现其有4个活泼氢质子信号没有显示出来,而根据相关化学位移可确定其是同一个已知化合物。13C-NMR (150 MHz, MeOD)显示有13个碳信号,结合DEPT谱可推测有6个芳香碳δC 123.4,119.4,126.2,128.2,152.5,115.6,2个羰基碳δC 171.8,172.4,而δC 162.1为醛基碳,2个次甲基碳δC 59.4,68.4,1个甲基碳δC 20.5,以及1个甲氧基碳δC 52.9。对其碳信号进行归属:δC 115.6 (C-1)、152.5 (C-2)、128.2 (C-3)、126.2 (C-4)、119.4 (C-5)、123.4 (C-6)、171.8 (C-7)、59.4 (C-8)、172.4 (C-9)、52.9 (C-10)、162.1 (C-11)、68.4 (C-12)、20.5 (C-13)。以上数据与文献[14]对比基本吻合,故确定为N-formylantimyic acid methyl ester。
化合物9:白色粉末状固体,ESI-MS显示准分子离子峰m/z 499 [M+H]+。1H-NMR (600 MHz, DMSO)显示有19个氢信号:δH 8.20 (1H, s, H-11),6.82 (1H, s, H-10),6.32 (1H, dd, J = 10.5, 1.3 Hz, H-3),5.01 (1H, m, H-7),2.99 (1H, dd, J = 2.5, 15.8 Hz, H-8),2.80 (1H, dd, J = 10.3, 15.6 Hz, H-8),2.58 (1H, m, H-4),1.66 (1H, m, H-5),1.65 (3H, s, 2-Me),1.27 (1H, m, H-6),1.25 (1H, m, H-5),1.06 (3H, d, J = 6.5 Hz, 4-Me)和0.95 (3H, d, J = 5.9 Hz, 6-Me)。而13C-NMR (150 MHz, DMSO)结合DEPT谱显示只有14个碳信号:δC 166.0 (C-1),126.8 (C-2),147.4 (C-3),30.7 (C-4),37.6 (C-5),35.1 (C-6),74.5 (C-7),24.0 (C-8),149.4 (C-9),122.9 (C-10),151.3 (C-11),12.7 (2-Me),21.1 (4-Me)和16.2 (6-Me),说明这个化合物可能是一个具有对称结构的二聚体,通过与文献[15]中化合物conglobatin A对比后发现两者波谱数据完全吻合,故最终确定为conglobatin A。
3. 讨论
自上世纪发现青霉素以来,微生物中活性次生代谢产物一直是药物先导化合物的重要来源之一,据统计1940年—2019年间,科学家从微生物中开发出293种治疗不同疾病的临床药物[16]。但随着研究的深入,很多微生物及其次生代谢产物存在被重复开发和提取分离的问题,加之多重耐药性的产生,迫使人们需要开拓新的制造药物的微生物来源[5-6],而其中极地微生物资源是珍贵而特殊的。来自极地海洋等特殊生态环境的生物往往具有比陆地生物更为丰富的代谢途径和功能基因簇,增加了产生结构新颖且功能独特的次生代谢物的可能性。极地生物以微生物和一些能适应极端条件的海洋生物为主,然而与已报道的大量极地微生物相比,鲜有微生物活性天然产物相关研究报道,因此,极地微生物极具研究价值[17-18]。
笔者以一株采自北极海域海绵共附生放线菌Streptomyces sp. LHW11-07为研究对象,从其发酵浸膏中分离得到9个单体化合物1~9,包括环二肽化合物1~5,倍半萜化合物6,核苷类化合物7,以及两个其他结构类型化合物8和9,其中化合物1和2是首次分离于Streptomyces放线菌,而这些化合物的生物活性还有待进一步探究;本研究进一步丰富了该属放线菌的化学多样性,同时,为高值化开发利用极地微生物这一国家战略资源提供了物质基础和理论依据。
据文献报道,化合物1对所测试的病原性细菌和真菌均具有一定的对抗作用[19],化合物2测试了4种肿瘤细胞均无明显的细胞毒性[20],化合物3对海胆Strongylocentrotus intermedius胚胎具有细胞毒活性[9],化合物5具有抗炎活性并对H1N1和RSV病毒有一定的杀伤作用[21],化合物7作为一种内源性嘌呤核苷,具有降低血压、抑制血小板聚焦、舒张血管、减慢心律等生理活性[22],而化合物9可抑制癌细胞株的增殖,在体外对Trypanosoma brucei brucei GUTat 3.1表现出抗锥虫体活性等[23]。
-
表 2 10批肾衰宁颗粒HPLC指纹图谱相似度
样品号 S1 S2 S3 S4 S5 S6 S7 S8 S9 S10 对照指纹图谱 S1 1.000 0.980 0.910 0.941 0.934 0.948 0.916 0.941 0.950 0.874 0.965 S2 0.980 1.000 0.934 0.934 0.932 0.945 0.951 0.946 0.978 0.867 0.973 S3 0.910 0.934 1.000 0.952 0.947 0.937 0.972 0.940 0.945 0.914 0.973 S4 0.941 0.934 0.952 1.000 0.990 0.984 0.951 0.971 0.923 0.940 0.986 S5 0.934 0.932 0.947 0.990 1.000 0.978 0.938 0.976 0.910 0.910 0.979 S6 0.948 0.945 0.937 0.984 0.978 1.000 0.954 0.987 0.931 0.923 0.986 S7 0.916 0.951 0.972 0.951 0.938 0.954 1.000 0.949 0.966 0.913 0.979 S8 0.941 0.946 0.940 0.971 0.976 0.987 0.949 1.000 0.928 0.890 0.980 S9 0.950 0.978 0.945 0.923 0.910 0.931 0.966 0.928 1.000 0.892 0.969 S10 0.874 0.867 0.914 0.940 0.910 0.923 0.913 0.890 0.892 1.000 0.937 对照指纹图谱 0.965 0.973 0.973 0.986 0.979 0.986 0.979 0.980 0.969 0.937 1.000  下载: 导出CSV
下载: 导出CSV
表 3 各成分线性关系
对照品 回归方程 r 线性范围(μg/ml) 橙皮苷 Y=2.391 8X–2.798 3 0.999 7 40~400 丹酚酸B Y=10.689X–5.578 3 0.999 8 10~100 大黄酚 Y=58.983X+121.99 0.999 9 7~350
下载: 导出CSV
表 4 加样回收率试验结果
成分 取样量(V/ml) 样品含量(m/μg) 加入量(m/μg) 测得量(m/μg) 回收率(%) 平均回收率(%) 橙皮苷 1.00 383.02 95.88 773.46 101.80 100.27 1.00 380.39 95.88 764.91 100.26 1.00 380.31 95.88 763.64 99.95 1.00 384.46 95.88 771.17 100.83 1.00 385.81 95.88 772.95 100.94 1.00 389.20 95.88 764.32 97.81 丹酚酸B 1.00 120.79 31.80 250.42 101.91 98.82 1.00 122.50 31.80 251.14 101.12 1.00 125.56 31.80 248.78 96.87 1.00 127.57 31.80 254.87 100.07 1.00 127.74 31.80 251.44 97.25 1.00 127.75 31.80 249.44 95.66 大黄酚 1.00 72.67 17.79 138.19 92.06 97.36 1.00 69.62 17.79 135.41 92.44 1.00 68.46 17.79 140.83 101.69 1.00 71.03 17.79 141.25 98.66 1.00 68.98 17.79 141.57 102.00 1.00 71.92 17.79 141.17 97.30
下载: 导出CSV
表 5 10批肾衰宁样品中3种成分的测定结果
药品批号 橙皮苷(μg/ml) 丹酚酸B(μg/ml) 大黄酚(μg/ml) 61103102 66.99 42.29 44.65 51103110 83.41 39.34 42.89 51103111 94.75 41.36 44.51 51103009 346 80.57 43.21 43.44 51103011 593 83.51 42.04 42.58 51103018 486 79.94 41.94 42.96 51103010 471 90.03 41.88 44.03 41103033 563 83.48 43.01 43.94 61103101 85,92 42.93 43.85 51103105 88.04 41.76 42.94
下载: 导出CSV
-
[1] 国家药典委员会. 中华人民共和国药典2015年版(一部)[S]. 2015: 1052-1053. [2] WANG S S, ZHANG J F, GUO M, et al. The efficacy of Shen shuaining capsule on chronic kidney disease: a systematic review and meta-analysis[J]. Evid Based Complement Alternat Med,2016,2016:7515413. [3] CHEN X J, GAO S H, RUAN M N, et al. Shen-Shuai-Ning granule decreased serum concentrations of indoxyl sulphate in uremic patients undergoing peritoneal Dialysis[J]. Biosci Rep,2018,38(5):BSR20171694. doi: 10.1042/BSR20171694 [4] 谢小洪, 黄鹤宁. 肾衰宁颗粒结合西医基础治疗慢性肾脏病4期疗效观察[J]. 中国处方药, 2014, 12(12):131. doi: 10.3969/j.issn.1671-945X.2014.12.113 [5] LIU W, WANG D M, LIU J J, et al. Quality evaluation of Potentilla fruticosa L. by high performance liquid chromatography fingerprinting associated with chemometric methods[J]. PLoS One,2016,11(2):e0149197. doi: 10.1371/journal.pone.0149197 [6] 李强, 杜思邈, 张忠亮, 等. 中药指纹图谱技术进展及未来发展方向展望[J]. 中草药, 2013, 44(22):3095-3104. doi: 10.7501/j.issn.0253-2670.2013.22.001 [7] 易智彪, 薄雯映, 许冬瑾, 等. 太子参HPLC指纹图谱研究[J]. 中草药, 2009, 40(8):1308-1310. doi: 10.3321/j.issn:0253-2670.2009.08.042 [8] 黄慧梅, 柳润辉. 中药复方药动学研究进展[J]. 药学实践杂志, 2014, 32(2):88-91. doi: 10.3969/j.issn.1006-0111.2014.02.003 [9] 李东晓, 桂雪虹, 陈为, 等. 通腑泻热灌肠液中大黄素及大黄酚含量测定[J]. 中药新药与临床药理, 2015, 26(3):372-377. [10] 周萍. 高效液相色谱法测定大明胶囊中大黄酚的含量[J]. 黑龙江科技信息, 2015(27):123. [11] 张小红, 肖雪, 梁洁, 等. HPLC法测定林芝地区丹参中丹参酚酸B含量[J]. 亚太传统医药, 2015, 11(11):45-46. [12] 谭丽荷. 陈皮四物片中橙皮苷的含量测定[J]. 中国当代医药, 2015, 22(9):88-90. [13] 姚荣成, 林琼芬, 张雯洁. HPLC法测定肾衰宁胶囊中大黄素与大黄酚的含量[J]. 中国药房, 2010, 21(36):3447-3449. [14] 杨柳, 张颖, 刘季田媛, 等. 牛膝各化学拆分组分指纹图谱研究[J]. 中医药信息, 2015, 32(6):16-19. [15] 胡志军, 陈建秋. HPLC测定不同基原陈皮药材中橙皮苷含量[J]. 中国实验方剂学杂志, 2012, 18(10):95-98. doi: 10.3969/j.issn.1005-9903.2012.10.027 期刊类型引用(12)
1. 马启刚,徐光红,高贵,程志坤. 不同剂量羟考酮对胃肠肿瘤手术患者术后致痛物质水平和组织灌注及肠道屏障的影响. 中国临床研究. 2024(08): 1214-1218 .  百度学术
百度学术2. 杨岳,彭俊波,韩锋. 羟考酮复合右美托咪定对甲状腺腔镜手术患者全身麻醉气管插管应激反应的影响. 中国药物应用与监测. 2024(05): 568-572 . 百度学术3. 杨芳. 舒芬太尼诱发呛咳反应的研究进展. 中国城乡企业卫生. 2023(01): 42-44 . 百度学术4. 李一男. 舒芬太尼在小儿疝气修补术中的不良反应发生情况及其影响因素分析. 中国医学创新. 2023(02): 124-127 . 百度学术5. 田岩,赵媛媛. 右美托咪定联合羟考酮在腹腔镜胃肠手术患者中的应用研究. 中外医疗. 2023(23): 100-104 . 百度学术6. 童鹏才,徐小凯,洪婷,吴沛琴. 右美托咪定辅助全身麻醉对妇科腹腔镜手术患者麻醉恢复期躁动的影响及风险预测模型构建. 药品评价. 2023(08): 1024-1027 . 百度学术7. 吴维强,姚泽宇,王学军. 羟考酮与舒芬太尼在老年股骨转子间骨折患者PFNA内固定术中的应用效果对比. 局解手术学杂志. 2022(06): 515-519 . 百度学术8. 张武华,张宇菲. 羟考酮联合丙泊酚与舒芬太尼联合丙泊酚在气道困难者中的应用. 中国卫生标准管理. 2022(11): 156-159 . 百度学术9. 邓国鹏,孙亮亮,孙能宏. 氢溴酸依他佐辛联合舒芬太尼对经腹腔镜胃癌根治术患者炎性因子及疼痛介质的影响. 现代医学与健康研究电子杂志. 2021(02): 32-34 . 百度学术10. 张迪,王珊珊,张洁,李燕爽. 腹腔镜术后麻醉苏醒期病人躁动防控措施的应用分析. 全科护理. 2021(30): 4262-4265 . 百度学术11. 杨振宇,周莉媛,钱家树,林学正. 低流量七氟醚复合舒芬太尼在盆腔炎性包块腹腔镜微创术中的麻醉作用. 中国妇幼保健. 2021(24): 5639-5643 . 百度学术12. 赵超,张潇,田皇华,王双华. 羟考酮复合舒芬太尼诱导在腹腔镜胆囊切除术中的麻醉效果观察. 名医. 2020(15): 70-72 . 百度学术其他类型引用(0)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 7658
- HTML全文浏览量: 1833
- PDF下载量: 25
- 被引次数: 12
