

 下载:
下载:
 下载:
下载:

-
氟马西尼(flumazenil,FMZ)作为中枢苯二氮䓬受体(CBZR)拮抗剂,具有高度特异性和选择性,通过拮抗苯二氮䓬类药物与其受体的结合,用于逆转其所致的中枢镇静作用[1]。11C-氟马西尼(11C-flumazenil,11C-FMZ)可通过PET/CT显像定量分析CBZR与γ氨基丁酸(GABA)受体、氯离子通道偶联形成的复合受体GABA/CBZR,用于癫痫病灶及遗传性舞蹈病的检查[2]。但是11C的半衰期只有20.38 min,制备结束后如不及时注射入受检者体内,每摩尔(mol)药液的放射性活度,即比活度会很快降低。11C-FMZ在体内经肝酶水解,部分转变为无CBZR拮抗活性的羧酸代谢物[3],见图1。不同比活度的11C-FMZ在体内代谢有无差异,未见文献报告。本研究通过测定不同比活度的11C-FMZ在体内的代谢变化,拟为11C-FMZ的临床合理应用提供一定依据。
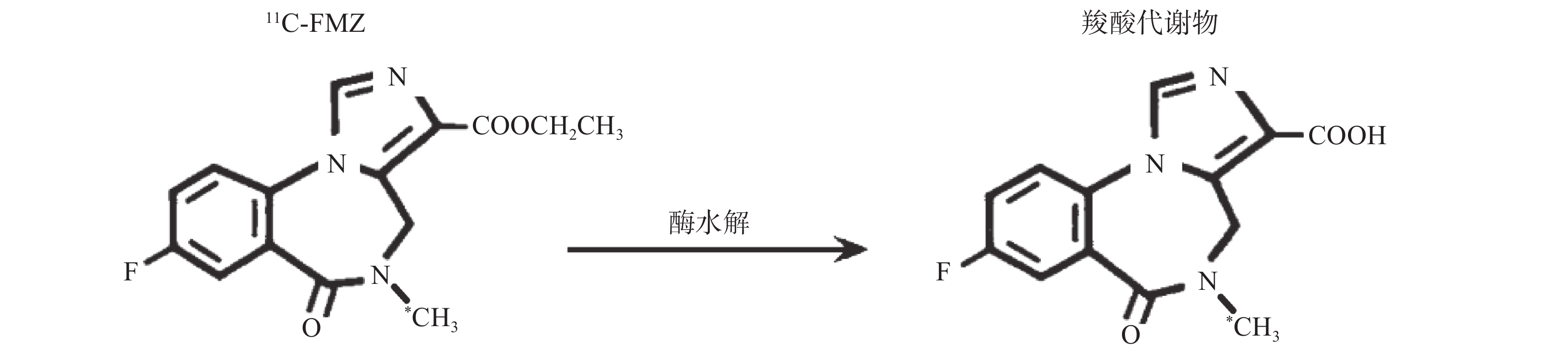
图 1 11C-FMZ及其水解产物
-
Minitrace回旋加速器、TRACELab FXc放射性药物合成系统(美国GE公司);HPLC半制备分析系统、紫外检测器、Sep-Pak C18层析柱(美国Waters公司);LB508型放射性检测器(德国Berthold公司);CRC-15R型活度检测器(美国Capintec公司);Millex-GS无菌滤膜(美国Capintec公司);Wallac 1470 WIZARD γ计数器(芬兰PE公司),WD-9405B型水平摇床(北京沃德生物医学仪器公司),高速低温离心机(美国Therma公司)。
去甲基氟马西尼、标准品氟马西尼(江苏华益科技有限公司);碘(美国Sigma-Aldrich公司);氢气、氦气、氮气、1%氧-氮混合气、盐酸、磷酸、无水乙醇、无水乙腈、二甲基甲酰胺(DMF)、氢化钠(NaH)、0.9%氯化钠(中国医药集团上海化学试剂公司);三氟磺酸银粉(Ag-Triflate,自制)。商品化溶剂皆为市售分析纯或化学纯。
-
选择2019年5月至10月北部战区总医院核医学科10名健康受试者,其中,男性5名,女性5名,平均年龄(41.7±4.7)岁,平均体重(69.3±6.8)kg。所有受试者均进行系统的体格检查,确认无基础疾病且试验前1个月内无药物服用史,并签署了知情同意书。
-
首先使用回旋加速器以1% O2/N2为原料,通过14N(p,α)11C核反应生产11C-CO2,传输至药物合成系统,并以图2所示,合成路线转化为11C-三氟甲基磺酰基甲烷(11C-Triflate-CH3)[4]。将0.5 mg去甲基氟马西尼和1 mg NaH溶于0.4 ml DMF于反应池中与11C-Triflate-CH3在0 ℃反应5 min,升至室温并加入0.5 mol/L HCl中和。混合物进入HPLC进行分离纯化,分离柱:Nucleosil 100-5 C18,250 mm×10 mm,流动相:含0.01 mol/L磷酸的22%乙腈溶液,检测波长254 nm,流速4 ml/min。收集放射性峰组分,用Sep-Pak C18柱再次分离纯化,最后通过0.22 μm的无菌滤膜过滤得到产物11C-FMZ[5],如图3所示。产品依据《中国药典》(2015年版)进行无菌检测和内毒素含量检测。

图 2 11C-Triflate-CH3合成路线
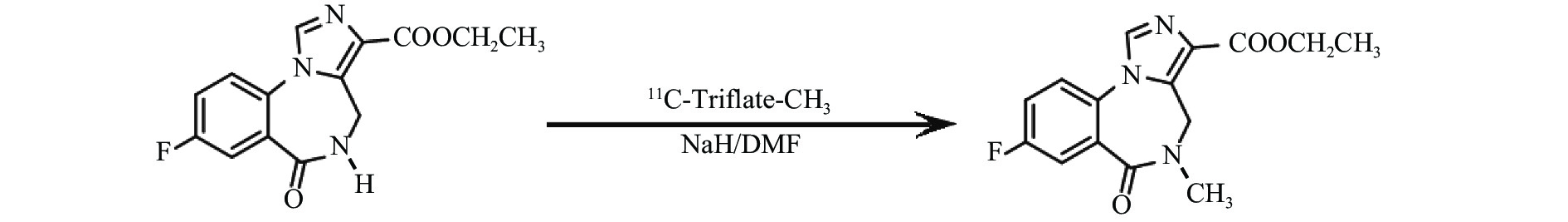
图 3 11C-FMZ合成路线
-
首先,在静息状态下通过肘静脉为10名受试者注射11C-FMZ 3 ml,经活度检测器测量,平均放射性活度为(10.5±2.9)mCi(1 Ci=3.7×1010 Bq),放射性比活度以单位物质的量所含放射性活度作为定量依据,平均比活度为(268.3±57.2)×103Ci/mol;2 h后,再次注射11C-FMZ 3 ml,平均活度为(10.6±2.8)mCi,此时药液已放置5~6个半衰期,平均比活度降为(57.8±11.4)×103Ci/mol。两次注射药物后均需静卧60 min,分别在1、2、3、5、10、15、20、30、40、60 min时于对侧肘静脉抽取血样。
-
取500 μl血样于试管中,加入500 μl缓冲溶液(0.1 mol/L硼酸,0.05 mol/L氯化钾,0.1 mol/L氢氧化钠)调节pH至11,再加入1 ml氯仿,于水平摇床混匀5 min,再以4 000 r/min离心5 min。取氯仿相和水相各200 μl,使用γ计数器分别测定两相1 min的放射性计数(counts per minute,cpm)。
-
放射性计数首先需经衰变公式(公式1)校正以抵消血浆预处理的耗时所致的放射性衰减,再计算各血样的百分注射剂量率(%ID/L,公式2)及不同比活度下各时间点11C-FMZ羧酸代谢物的占比(公式3)。使用SPSS 18.0软件对数据进行统计学分析,符合正态分布的计量资料以(
$ \stackrel{-}{x}\pm s $ )表示,采用配对样本均数t检验,P<0.05为差异有统计学意义。$$ {I}_{0}=\frac{I}{{e}^{-\lambda t}} $$ (公式1) I0:抽血时的cpm,I:测量时的cpm,t:抽血与测量的时间差 (min),T:11C半衰期20.38 min。
首先计算t/T值,从“衰变计算表”[6]中查出比值对应的e-λt值。
$$ {\text{百分注射剂量率}}\left( {{\rm{\% ID}}/{\rm{L}}} \right) = \frac{{{\text{血浆的放射性计数}}\left( {{\rm{cpm}}} \right)}}{{{\text{血浆体积}}\left( {\rm{L}} \right)}} $$ (公式2) $$\begin{split} &{\rm{{\text{羧酸代谢物的占比}}}}\left( {\rm{\% }} \right) =\\ & \frac{{{\text{羧酸代谢物的}}{\rm{\% ID}}/{\rm{L}}}}{{{}^{11}{\rm{C}} - {\rm{FMZ}}{\text{的}}\% {\rm{ID}}/{\rm{L}} + {\text{羧酸代谢物的}}{\rm{\% ID}}/{\rm{L}}}} \end{split}$$ (公式3) -
11C-FMZ的百分注射剂量率随时间呈指数下降,羧酸代谢物则先升后降并在10 min后开始超过11C-FMZ,如表1所示。羧酸代谢物的百分占比随时间延长而逐步增加,于15 min后基本趋于稳定,且低比活度组的百分占比明显高于高比活度组,从统计学结果可以看到,两组数据在15~60 min的区间存在显著统计学差异(P<0.05),如表2所示。
表 1 不同比活度11C-FMZ注射后在不同时间的百分注射剂量率(%ID/L)
测定成分 时间(t/min) 1 2 3 4 5 10 15 20 30 40 60 11C-FMZ① 9.76±4.80 6.00±1.42 3.30±0.60 2.77±0.39 2.25±0.33 1.95±0.24 1.55±0.15 1.28±0.18 1.09±0.14 0.98±0.15 0.78±0.10 羧酸代谢物① 0.04±0.04 0.19±0.10 0.44±0.13 0.51±0.14 0.67±0.13 1.88±0.24 2.00±0.33 1.73±0.28 1.50±0.28 1.59±0.20 1.22±0.22 11C-FMZ② 13.84±4.67 6.10±1.31 3.27±0.49 3.10±0.36 2.66±0.37 1.97±0.33 1.42±0.13 1.23±0.15 1.11±0.06 0.67±0.13 0.45±0.07 羧酸代谢物② 0.15±0.11 0.28±0.08 0.50±0.17 0.55±0.29 0.98±0.27 2.22±0.42 2.36±0.44 2.39±0.34 1.90±0.41 1.39±0.25 0.98±0.14 注:①表示高比活度(268.3±57.2)×103 Ci/mol注射后的剂量率;②表示低比活度(57.8±11.4)×103Ci/mol注射后的剂量率。 表 2 不同比活度11C-FMZ的羧酸代谢物在不同时间百分注射剂量率的占比(%)
11C-FMZ羧酸代谢物 时间(t/min) 1 2 3 4 5 10 15 20 30 40 60 高比活度 0.54±0.54 3.32±1.89 12.04±3.61 15.58±4.25 23.11±4.92 48.98±4.89 56.11±4.68 57.36±5.63 57.60±3.45 61.92±4.51 60.85±5.39 低比活度 1.12±0.70 4.53±1.69 13.48±4.94 14.64±6.01 27.04±7.81 52.90±5.81 62.04±4.69* 65.85±5.35* 62.54±5.75* 67.26±4.88* 68.26±4.47* *P<0.05,与高比活度组比较。 -
本研究使用氯仿作为11C-FMZ及其羧酸代谢物的分离试剂,对11C-FMZ的提取效率可达100%,而代谢物可完全保持在水相中。相关研究中,TLC没有检测到除羧酸代谢产物以外的其他放射性代谢物[7],保证了该方法对放射性比活度测定的准确性。
Debruyne等[8]研究了进食对11C-FMZ体内代谢的影响,并观察到进食后的代谢率更高,并认为是进食导致肝脏血流增加,从而导致肝脏提取量高的药物代谢增加。由于11C-FMZ主要通过肝脏代谢从体内清除,仅0.2%的给药剂量在尿中以原形排出[9],因此在PET研究中,应控制饮食条件。
每升血浆中所含的标记药物的百分比与之前的某些定量研究结果基本一致。Samson等[10]和Shinotoh等[11]对高放射性比活度研究的结果分别为10 min时(2.4±0.9)和(3.7±1.6)%ID/L,20 min时(2.1±0.4)和(3.5±1.4)% ID/L。但二者均缺少低放射性比活度及羧酸代谢物的进一步研究。本研究中血浆的百分注射剂量率在注射后10 min左右出现上升,与之前相关研究结果相似[11-13],这可能与标记的羧酸代谢物的产生有关。Klumpers等认为原因是药物在分布阶段暴露于酯酶的某种首过效应,这种酯酶在肝脏、肾脏和大脑中大量存在且具有非常广泛的底物特异性[14]。在狒狒身上进行的相关研究没有观察到该现象[8],这种差异是由于在该物种中羧酸代谢物的标记分数较低。本研究中的高、低比活度对应代谢物的占比分别为61%和68%,而狒狒仅为45%。
综上所述,放射性比活度不同的11C-FMZ注射后的代谢率存在明显差异,在PET相关研究中应尽量避免对比活度差异过大的同一批受试者进行试验,临床应用时同一患者在复查时所采用的比活度也需尽量与初诊时一致,以获得最佳的图像对比价值。
The metabolic analysis of 11C-flumazenil in different specific radioactivity
-
摘要:
目的 通过测定11C-氟马西尼(11C-flumazenil,11C-FMZ)在体内主要代谢物的百分占比,分析不同比活度11C-FMZ的代谢差异。 方法 选取2019年5月至10月10名受试者,其中,男性5名,女性5名,平均年龄(41.7±4.7)岁,平均体重(69.3±6.8)kg。先后注射高低两种比活度(268.3±57.2)×103和(57.8±11.4)×103Ci/mol的11C-FMZ,并分别测定注射后1、2、3、4、5、10、15、20、30、40、60 min时血浆中11C-FMZ及其代谢物的百分注射剂量率,使用配对样本均数t检验计算并对比两组代谢物的百分占比。 结果 代谢物的百分占比随时间逐步增加,于15 min后基本趋于稳定,且低比活度组的百分占比明显高于高比活度组,在15~60 min的区间存在显著统计学差异(P<0.05)。 结论 不同比活度的11C-FMZ注射后的代谢率存在明显差异,临床应用时应尽量避免比活度差异过大。 Abstract:Objective To analyze the metabolic differences of 11C-flumazenil (11C-FMZ) with different specific radioactivity by detecting the percentage proportion of the main metabolites in vivo. Methods 5 male and 5 female volunteers with average age of (41.7±4.7) years and weight of (69.3±6.8) kg were selected from May to October 2019. 11C-FMZ with high and low specific radioactivity (268.3±57.2)×103 and (57.8±11.4)×103 Ci/mol was injected successively. The percentage of injected dose/liter of 11C-FMZ and its metabolites in the plasma at 1, 2, 3, 4, 5, 10, 15, 20, 30, 40 and 60 min after the injection was measured. Paired sample mean t test was used to calculate and compare the percentage of metabolites in the two groups. Results The percentage proportion of metabolites increased gradually with time, and reached the stable level after 15 min. The percentage proportion of low specific radioactivity group was higher than that of high specific radioactivity group with a significant statistical difference between 15 min and 60 min (P<0.05). Conclusion The metabolic rate of 11C-FMZ with different specific radioactivity was significantly different after injection and the specific radioactivity difference should be avoided if possible in clinical application. -
Key words:
- 11C-flumazenil /
- carboxylic acid metabolites /
- specific radioactivity
-
补体系统是人体重要的免疫防御系统之一,是由30多种广泛存在于血清、组织液和细胞膜表面的蛋白质组成的,具有精密调控机制的蛋白质反应系统,其主要通过3种途径激活:经典途径、旁路途径和甘露糖结合凝集素途径。补体系统正常激活,可在靶细胞上形成膜攻击复合物,导致靶细胞的溶解,补体的这一功能在机体的免疫系统中起重要的防御和免疫监视作用,对抵御外来微生物的入侵和维持机体平衡有重要的作用。然而该系统的过度激活将释放炎性过敏毒素C3a和C5a,具有化学诱导作用的C5a能趋化嗜中性粒细胞、中核细胞和嗜酸性粒细胞,这些细胞释放蛋白酶和具有趋化作用细胞因子,进一步聚集T、B淋巴细胞和其他炎性细胞,从而促进炎症反应的发生,引起系统性红斑狼疮、类风湿性关节炎、动脉粥样硬化、肾小球肾炎等[1-2]。近年来已有研究表明[3],补体系统的激活是类风湿性关节炎中慢性滑膜炎的发病因素之一。因此,抑制补体系统的过度激活可能是治疗类风湿性关节炎的重要机制之一。
三色片为复旦大学附属中山医院的院内制剂,由雷公藤、黄芪和丹参三味药材按1∶1∶1的比例配伍组成,在临床上用于治疗类风湿性关节炎、系统性红斑狼疮、银屑病和湿疹等结缔组织疾病。我院临床医生在长期的医疗实践中总结出来的经验方,效果显著[4]。组方中雷公藤,性味辛寒,有大毒,归肝、肾经,具有清热解毒、活血化瘀、通络止痛、杀虫止痒等功效。现代研究表明,雷公藤内酯醇对大鼠脑皮质内注射β-淀粉酶后补体C1q和C3的表达有抑制作用,表明雷公藤对补体系统有抑制作用,目前临床上广泛用于治疗类风湿性关节炎、系统性红斑狼疮、银屑病和湿疹等结缔组织疾病[5]。组方中的黄芪用于脾肺气血或中气下陷之症、卫气虚所致表虚自汗、气虚血滞导致的肢体麻木、关节痹痛等症,可联合治疗类风湿性关节炎[6]。黄芪在治疗2型糖尿病大鼠的研究中发现其能降低补体C3的水平,表明其对补体系统具有一定的调节作用[7-8]。丹参是最常用的活血化瘀中药之一,具有祛瘀止痛,养血安神的功效,现代药理学研究表明其还具有保护肝脏的功能[9],可拮抗雷公藤的肝毒性。本研究通过经典途径抗补体活性测定方法筛选出三色片醇提物的乙酸乙酯部位抗补体活性最佳,并采用UPLC-Q-TOF-MS法对该部位的化学成分进行结构表征,为三色片抗补体活性药效物质基础及治疗补体过度激活相关疾病提供科学依据。
1. 仪器、试剂与材料
Tripie TOF5600+型四级杆-飞行时间串联质谱仪,配备电喷雾电离源和CDS自动校正系统(美国Applied Biosystems公司);Peak view2.2和Master view1.1数据处理系统(美国Applied Biosystems公司);LC-30A超高效液相色谱仪,包括高压输液泵,自动进样器,柱温箱和在线脱气机(日本岛津公司);KQ5200E型超声清洗器(昆山市超声仪器有限公司);甲醇、乙腈(色谱纯,德国Merck公司);甲酸(色谱纯,美国Sigma-Aldrich公司); 蒸馏水(娃哈哈集团);三色片提取物由作者自制,现样品存放于复旦大学附属中山医院药剂科(SSP2018);补体、溶血素(自制);毛蕊异黄酮(批号:ST088101),雷公藤甲素(批号:ST020501),雷公藤内酯酮(批号:ST049901),丹参酮II A(ST014601)、黄芪甲苷(ST001601)(纯度≥ 98%,均购自上海斯丹德生物技术有限公司)。
2. 方法
2.1 三色片醇提物及各极性部位的制备
雷公藤、黄芪和丹参三味药材按1∶1∶1配伍,其中,黄芪和丹参加6倍量的水浸泡2 h后,煎煮2次,第一次1.5 h,第二次加水4倍量煎煮1 h,煎液滤过,合并滤液并浓缩至相对密度为1.10~1.20(70 ℃),加入2倍量的乙醇,静置沉淀24 h,取上清液备用。雷公藤分别加4倍量的乙醇加热回流2次,每次1.5 h,合并提取液,滤过,加入上述备用药液,混匀,回收乙醇至无醇味,浓缩后即得三色片醇提物,经现有的质量标准检验为制备三色片制剂合格的提取物。精密称取三色片醇提物2.0 g,置于100 ml萃取瓶中,加25 ml蒸馏水溶解后,用等量的石油醚、乙酸乙酯和正丁醇进行萃取,浓缩干燥后,放冷至室温,得到三色片醇提物的石油醚部位0.36 g,乙酸乙酯部位0.42 g,正丁醇部位0.56 g和水溶性部位。
2.2 经典途径的抗补体活性测定
取各极性部位样品2 mg溶于DMSO,采用BBS缓冲液稀释成不同浓度的样品,并加入临界浓度的补体(1∶80稀释的豚鼠血清),溶血素和2%绵羊红细胞(SRBC)。37 ℃水浴30 min,离心后取上清液在405 nm波长下测定吸光度(A)值。同时设置中药对照组(将等量的中药提取物加入BBS缓冲液中,用于测定中药本底A值)、补体组(取临界浓度的补体直接加入适量的BBS缓冲液、溶血素和2%SRBC,用于测定临界浓度补体所造成红细胞溶血的A值)和全溶血组(将2%SRBC加入水中使之全溶血,用于观察补体组是否达到或接近全溶血水平),并以肝素作为阳性对照组,计算溶血抑制率。以供试品浓度为横坐标(X),溶血抑制率为纵坐标(Y),计算CH50(经典途径50%抑制溶血所需供试品浓度)。溶血抑制率=1−(A中药−A中药对照)/A全溶血。
2.3 不同浓度的样品色谱与质谱条件
2.3.1 色谱条件
色谱柱为ACQUITY UPLC BEH C18(2.1 mm×100 mm,1.7 μm);流动相0.1%甲酸和水溶液(A)−乙腈(B);梯度洗脱:0~9 min,10%~23% B;9~13 min,23% B;13~28 min,23%~40% B;28~32 min,40%~50% B;32~37 min,50%~100% B;37~42 min,100% B;42~42.1 min,10%B;42.1~50 min,10% B;流速为0.25 ml/min,柱温为35 ℃;进样量为2 μl。
2.3.2 质谱条件
在正/负离子模式,离子源选择电喷雾离子化源(ESI);使用m/z 50~1250扫描范围;碰撞能量35 eV,碰撞能量叠加(35±15)eV;喷雾电压5 500 V;雾化气温度550 ℃;去簇电压100 V;雾化气和辅助气均为50 psi;气帘气25 psi;数据采集时间50 min;采用母离子触发的子离子(TOF-MS-IDA-MS/MS)扫描方式;多重质量亏损和动态背景扣除为触发二级的条件,满足该条件进行二级扫描。
2.4 对照品溶液的制备
精密称取毛蕊异黄酮、雷公藤甲素、雷公藤内酯酮、丹参酮Ⅱ A和黄芪甲苷对照品1.0 mg,加甲醇2 ml,溶解,摇匀,即得各对照品溶液。
2.5 供试品溶液的制备
取三色片醇提物的乙酸乙酯部位样品0.2 g,置于10 ml量瓶中,加入70%甲醇5 ml,超声处理(功率250 W,频率40 kHz)30 min,放冷至室温,70%甲醇定容至刻度,摇匀,滤过,取续滤液,即得供试品溶液。
2.6 三色片中化学成分数据库的建立
根据三色片中各药材化学成分研究文献,收集3种药材所含化合物成分的基本信息,包括化合物名称、分子式、精确分子量、准分子离子峰和碎片离子峰。通过精确分子量匹配,对照品的保留时间,二级谱所得到的离子碎片与文献报道进行比对,最终确定化合物的结构。
3. 结果与分析
3.1 三色片醇提物各极性部位的抗补体活性
分别对三色片醇提物的石油醚部位、乙酸乙酯部位和正丁醇部位进行经典途径的抗补体活性测定,以肝素为对照品,结果发现乙酸乙酯部位的抗补体活性最好,其抗补体活性略低于肝素钠,其次是正丁醇部位,结果见表1。
表 1 三色片提取物不同部位抗补体活性测定编号 研究对象 抗补体活性(CH50,μg/ml) 1 肝素 14.4±1.2 2 三色片-石油醚部位 − 3 三色片-乙酸乙酯部位 233.9±10.1 4 三色片-正丁醇部位 344.0±14.5 注:“—”表示该部位无抗补体活性。 3.2 三色片醇提物乙酸乙酯部位的UPLC-Q-TOF-MS分析
精密吸取对照品溶液和供试品溶液2 μl,采用“2.1”项下的色谱与质谱条件对样品进行分析,通过正、负离子全扫描,获得正、负离子模式下的总离子流图,见图1。
通过与对照品比对,分子离子峰质谱数据解析,与参考文献比对,共鉴定出三色片醇提物乙酸乙酯部位42个化合物,结果见表2。
表 2 三色片提取物中各成分主要碎片离子及谱峰归属化合物
编号tR/min 分子式 理论值(m/z) 模式 实测值(m/z) 误差(×10−6) 碎片离子(m/z) 化合物名称 参考文献 1 3.54 C7H6O3 139.039 0 [M+H]+ 139.039 4 3.0 121.028 7 原儿茶醛 [10] 2 4.76 C21H27N3O3 370.212 5 [M+H]+ 370.214 3 4.8 249.124 6,160.112 6,95.013 3,166.086 6,100.076 2,91.054 8 南蛇藤糠酰胺碱 [11] 3 6.63 C22H22O10 447.128 6 [M+H]+ 447.130 4 4.2 285.077 5,270.053 5,253.050 8,225.055 6,137.023 5 毛蕊异黄酮-7-O-β-D-葡萄糖苷 [12] 4 7.04 C23H29N3O2 380.233 3 [M+H]+ 380.235 1 4.8 176.106 9,160.112 6,105.033 8,100.076 5 苯代南蛇碱 [11] 5 9.41 C9H10O5 197.045 6 [M-H]− 197.044 7 −4.2 179.038 3,135.044 3 丹参素 [10] 6 9.42 C9H8O4 179.035 0 [M-H]− 179.034 2 4.7 135.044 8 咖啡酸 [13] 7 11.16 C16H12O4 431.133 7 [M+H]+ 431.136 3 6.1 269.082 6,253.050 3,225.055 5,213.091 7,197.060 2,136.014 6,118.041 7 芒柄花苷 [12] 8 12.71 C16H12O5 285.075 8 [M+H]+ 285.077 4 5.7 270.053 4,253.050 3,225.055 3,137.023 5 毛蕊异黄酮* [12] 9 12.76 C17H16O5 301.107 1 [M+H]+ 301.109 0 6.4 167.070 8,152.047 3,147.043 2,105.034 0,123.043 3 astrapterocarpan [12] 10 13.06 C20H24O6 361.164 6 [M+H]+ 361.166 5 5.5 269.154 3,227.108 3,185.096 9,157.101 7,129.070 3,91.054 9 雷公藤甲素* [14-15] 11 13.92 C17H14O6 315.086 3 [M+H]+ 315.088 1 5.6 300.064 7,243.065 5,167.034 2 熊竹素 [16] 12 18.21 C18H12O7 341.065 6 [M+H]+ 341.066 9 3.9 295.060 7,277.050 9,249.056 0 丹酚酸G [17] 13 19.76 C26H20O10 491.098 4 [M-H]− 491.097 0 −2.9 311.054 9,293.044 6,267.064 6,135.044 7 丹酚酸C [18] 14 21.46 C16H12O4 269.080 8 [M+H]+ 269.082 7 4.1 253.015 3,237.052 6,225.055 5,213.092 3,136.015 9,118.041 7,197.060 2 芒柄花素 [12] 15 22.57 C36H45NO17 764.276 0 [M+H]+ 764.278 3 2.9 746.276 1,686.246 3,644.235 1,206.081 7,188.070 9,178.086 5 aquifoliunine E-Ⅲ [14] 16 23.45 C20H22O6 359.148 9 [M+H]+ 359.150 7 4.9 267.138 0,225.019 5,183.079 9,128.061 8,91.054 3 雷公藤内酯酮* [19] 17 24.01 C38H47NO19 822.281 5 [M+H]+ 822.284 1 3.2 804.275 8,204.066 2,176.071 4 alatusinnine [20] 18 25.01 C39H45NO19 832.265 9 [M+H]+ 832.269 0 3.8 804.273 3,194.081 9,176.071 2 hypoglaunine E [11] 19 26.14 C41H68O14 829.458 0 [M+COOH]− 829.460 7 3.3 783.457 9,621.404 3,489.357 2 黄芪甲苷* [14] 20 28.19 C38H47NO18 806.286 6 [M+H]+ 806.290 3 3.8 788.279 5,686.247 0,206.082 1, 178.086 5 雷公藤定宁 E [20] 21 28.65 C39H45NO18 816.271 0 [M+H]+ 816.273 9 3.6 798.261 9,756.250 9,206.081 3,178.086 1,160.075 2 1-去乙酰基雷公藤吉碱 [11] 22 28.70 C43H70O15 871.468 6 [M+COOH]− 871.470 8 2.5 825.470 2,765.448 2,489.356 8 黄芪皂苷Ⅱ [13] 23 29.09 C41H47NO20 874.276 4 [M+H]+ 874.278 5 2.3 856.269 2,846.282 9,828.272 3,674.245 1,204.065 6,176.070 7 雷公藤春碱 [11] 24 29.72 C38H47NO18 806.286 6 [M+H]+ 806.291 2 3.8 788.280 4,686.247 4,206.082 4 peritassine A [20] 25 30.24 C43H70O15 871.468 6 [M+COOH]− 871.470 3 2.5 825.464 1,765.440 5 异黄芪皂苷Ⅱ
(异构体1)[16] 26 30.89 C19H16O4 309.112 1 [M+H]+ 309.114 2 6.7 281.667 0,263.106 0,235.076 7 丹参醛 [21] 27 31.58 C43H70O15 871.468 6 [M+COOH]− 871.470 8 2.5 825.470 2,765.448 0 异黄芪皂苷Ⅱ
(异构体2)[13] 28 32.12 C21H20O4 337.143 4 [M+H]+ 337.142 5 −2.7 309.686 6 丹参新醌丁 [10] 29 32.16 C43H49NO19 884.297 2 [M+H]+ 884.299 7 2.8 856.304 5,674.246 0,204.663 0,176.071 2 雷公藤定碱 [14] 30 32.96 C45H72O16 913.479 1 [M+COOH]− 913.482 4 3.5 867.481 7,825.469 8,807.464 3,765.450 6 黄芪皂苷Ⅰ [16] 31 32.99 C41H47NO19 858.281 5 [M+H]+ 858.285 4 4.6 840.275 7,798.263 8,746.269 1,738.243 5,686.248 0,206.082 5,178.087 1 雷公藤晋碱 [20] 32 33.04 C38H47NO18 806.286 6 [M+H]+ 806.289 7 3.8 788.278 3,686.244 4,206.082 1,728.257 0 卫矛碱 [20] 33 33.70 C45H72O16 913.479 1 [M+COOH]− 913.484 7 3.8 867.478 5,825.283 5,807.458 4,765.432 6 异黄芪皂苷Ⅰ
(异构体1)[16] 34 34.60 C45H72O16 913.479 1 [M+COOH]− 913.483 3 3.8 867.477 8,825.282 1,807.456 4,765.443 2 异黄芪皂苷Ⅰ
(异构体2)[16] 35 34.92 C46H49NO22 968.281 9 [M+H]+ 968.286 3 4.5 856.677 0,838.257 4,684.228 8,204.065 6,178.070 8 雷公藤素B [20] 36 35.01 C43H49NO18 868.302 2 [M+H]+ 868.304 6 2.7 868.364 0,850.295 8,746.268 9,686.247 6,206.082 4,178.087 1 雷公藤次碱 [20] 37 35.02 C41H47NO17 826.291 7 [M+H]+ 826.295 1 4.2 808.285 3,748.264 0,206.082 2,178.086 8 tripterygiumine Ⅰ [20] 38 35.49 C19H20O3 297.148 5 [M+H]+ 297.145 0 4.8 251.144 0,279.139 3,254.054 9,268.110 5,282.126 3 隐丹参酮 [10,17] 39 35.70 C20H28O2 299.201 7 [M-H]− 299.199 6 −6.7 283.168 2,213.090 8,201.916 0, 雷酚萜 [22] 40 35.86 C48H51NO18 930.317 9 [M+H]+ 930.321 3 3.7 912.308 7,310.111 0,206.081 8,188.071 2,178.086 5,105.033 6 ebenifoline E-Ⅱ [20] 41 36.81 C19H18O3 295.132 9 [M+H]+ 295.134 9 4.0 277.124 3,249.127 5,266.095 3,262.097 7,280.109 9 丹参酮Ⅱ A* [10,18,23] 42 37.49 C19H22O2 283.169 3 [M+H]+ 283.169 3 0 265.098 1,240.032 2,223.106 7,195.095 8,181.101 1 丹参新酮 [17,21,24] 注:*表示与对照品鉴定的化合物。 3.2.1 黄酮类化合物结构解析
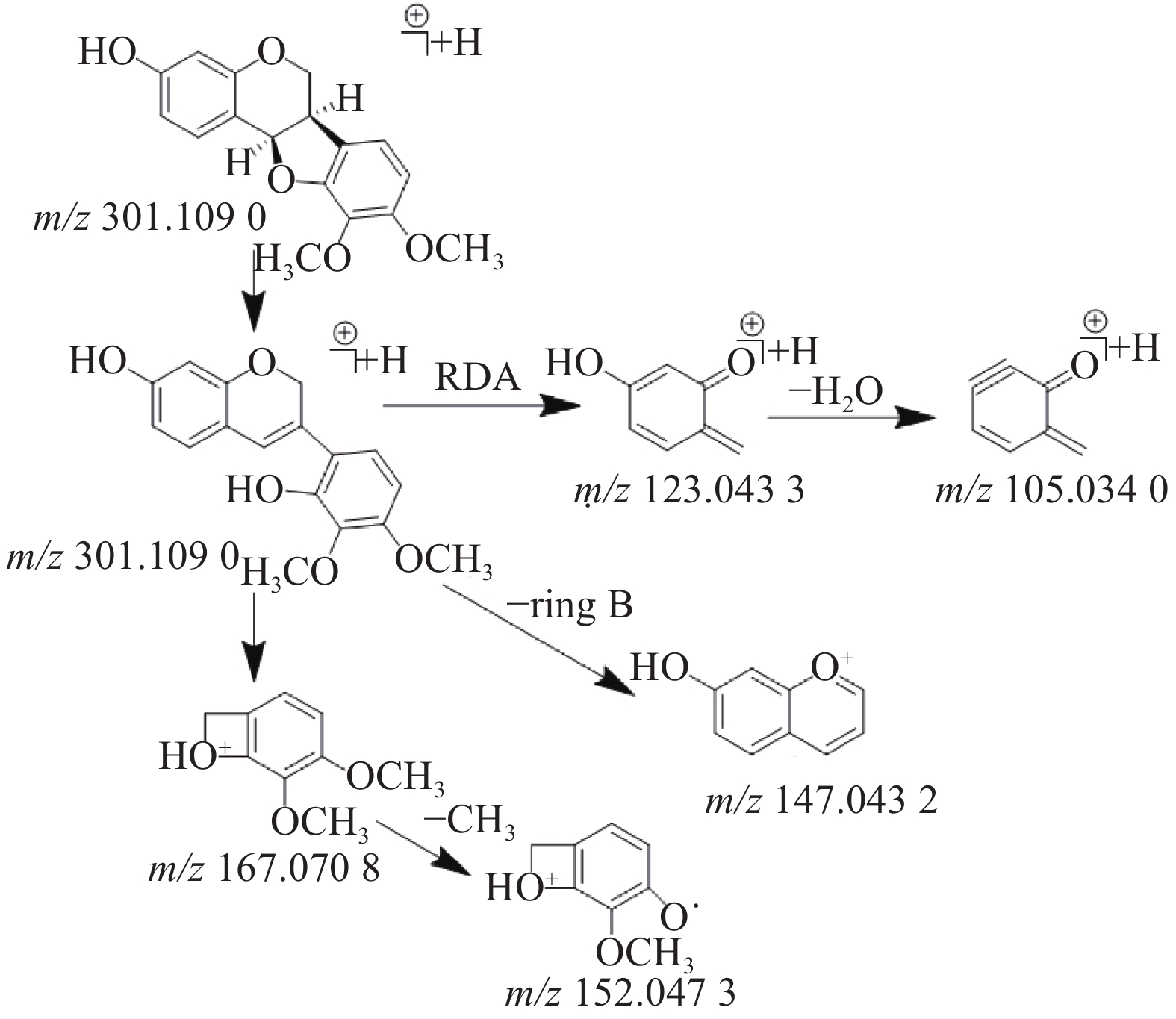
在乙酸乙酯部位中共鉴定出6个黄酮类化合物,其中4个黄酮苷元和2个黄酮苷,苷元为黄酮、异黄酮和紫檀烷,该类化合物在正离子模式下具有较好的响应。二级质谱中黄酮苷元易发生中性丢失,形成[M+H-H2O]+、[M+H-CO]+、[M+H-CH3]+等碎片离子,如在化合物8的二级质谱中可见m/z 270.053 4和m/z 253.050 3,则为m/z 285.077 4分别脱去-CH3和CH3OH形成的[M+H-CH3]+和[M+H-CH3OH]+碎片离子峰,m/z 225.055 3是m/z 253.050 3脱去1分子的CO形成的碎片离子峰,通过对照品的保留时间和参考文献[12]质谱数据比对确定化合物8为毛蕊异黄酮,m/z 137.023 5的碎片离子峰为异黄酮母核C环发生RDA裂解所产生。黄酮苷类易脱去糖基形成较强的分子离子峰,如化合物3(m/z 447.130 4)的二级质谱脱去糖基形成m/z 285.077 5的分子离子峰,并与化合物8(m/z 285.077 4)的二级质谱图非常相似,说明化合物3和化合物8在结构上是相似的,但化合物3的分子量多了162(C6H10O5),通过数据库比对和参考文献[12]推测化合物3则为毛蕊异黄酮-7-O-β-D-葡萄糖苷。化合物9(m/z 301.109 0)通过数据库比对发现两种候选化合物分别为astrapterocarpan和astraisoflavan,二级质谱中主要碎片离子峰为C环裂解产生的含A环和B环片段的碎片离子,其中,m/z 167.070 8为含B环的碎片离子峰且为基峰,进一步脱甲基形成m/z 152.047 3,m/z 123.043 3为含A环的碎片离子峰,进一步脱水形成m/z 105.034 0,m/z 147.043 2为母离子m/z 301.109 0脱去B环形成的碎片离子峰,根据m/z 167.070 8的碎片离子峰为基峰和含有m/z 147.043 2的碎片离子峰这两个特征,结合参考文献[12]的质谱数据,推测该化合物为astrapterocarpan,其相关裂解途径见图2。
3.2.2 三萜皂苷类化合物结构解析
在乙酸乙酯部位中鉴定出7个三萜皂苷类化合物,在负离子模式下均具有较好的响应,一级质谱中产生[M+COOH]−的准分子离子峰,二级质谱中产生较强的[M-H]-碎片离子峰和脱去糖基的较弱的分子离子峰。化合物19在负离子模式下产生的准分子离子峰为[M+COOH]−(m/z 829.458 0),二级质谱中产生m/z 783.457 9[M-H]−峰,脱去1分子六碳糖(C6H10O6)形成m/z 621.404 3的碎片离子峰,m/z 489.357 2则为m/z 621.404 3进一步脱去1分子五碳糖(C5H6O5)后形成的苷元碎片离子峰,推测其苷元为9,19-环阿尔廷烷,通过对照品的保留时间,参考文献[13]的离子碎片比对确定该化合物为黄芪甲苷。
3.2.3 生物碱类化合物结构解析
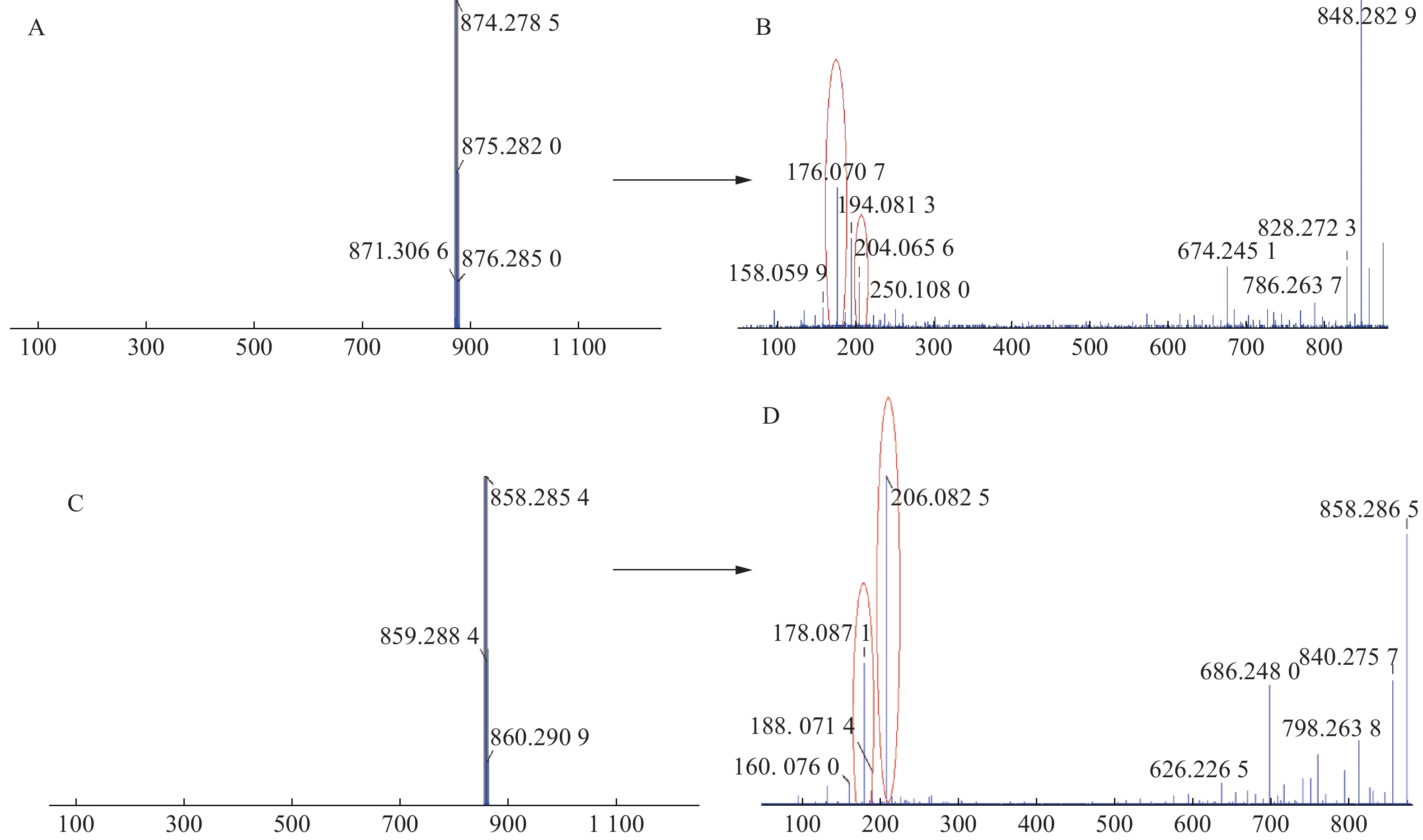
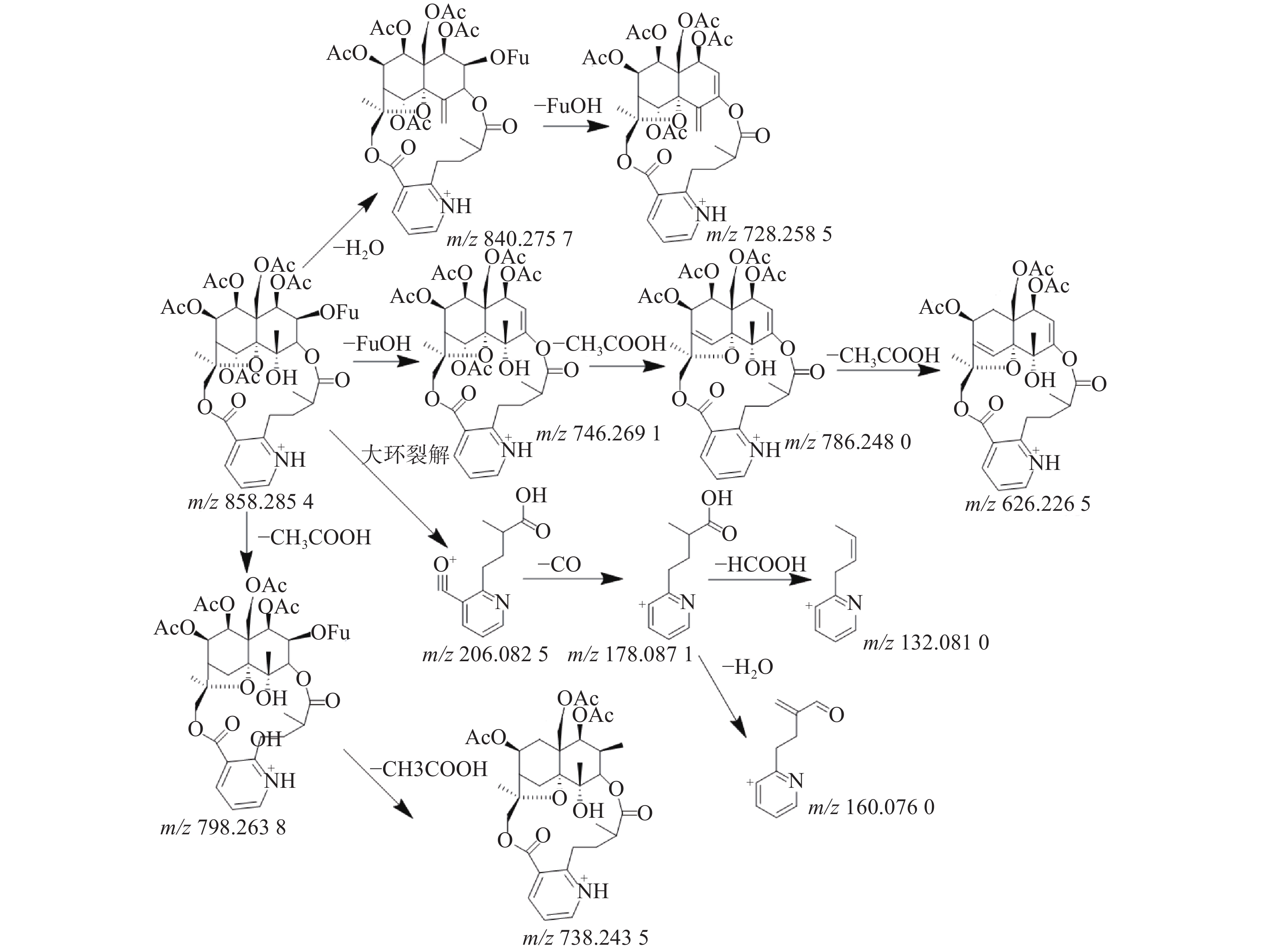
三色片提取物中共鉴定出16个生物碱类化合物,均来自雷公藤药材,在正离子模式下具有较好的响应,一级质谱中产生[M+H]+的准分子离子峰,二级质谱发现该类型的化合物容易脱去H2O、CO和CH3COOH等中性小分子而产生碎片离子峰,多数生物碱含有吡啶二羧酸部位的碎片离子峰。如化合物23在正离子模式下产生m/z 874.278 5的准分子离子峰,二级质谱中产生脱去1分子CO的m/z 846.282 9的基峰,脱去1分子H2O的m/z 856.2692的碎片离子峰和脱去1分子HCOOH的m/z 828.272 3的碎片离子峰,m/z 674.245 1峰为m/z 846.2829脱去C5H4O3侧链和CH3COOH形成的碎片离子峰,m/z 204.065 6峰为大环开裂产生的吡啶二羧酸部分脱水产生的碎片离子,该离子进一步脱羧形成m/z 176.070 7的碎片离子,通过数据库和参考文献[11]质谱数据的比对,推测化合物23为雷公藤春碱。化合物31在正离子模式下产生m/z 858.285 4的准分子离子峰,二级质谱中产生脱去1分子H2O的m/z 840.275 7的碎片离子峰,准分子离子峰脱去1分子CH3COOH形成较强的m/z 798.263 8峰,在进一步脱去1分子CH3COOH形成738.243 5峰,准分子离子峰m/z 858.285 4脱去FuOH(C5H4O3)侧链形成的m/z 746.269 1的碎片离子峰,再进一步脱去1分子CH3COOH,形成m/z 686.248 0的碎片离子,m/z 206.082 5峰为大环开裂产生的吡啶二羧酸部分脱水产生的碎片离子,该离子进一步脱羧形成m/z 178.087 1的碎片离子,通过数据库和参考文献[20]质谱数据的比对,推测化合物31为雷公藤晋碱。雷公藤晋碱中吡啶二羧酸部分较雷公藤春碱中少一个羟基,故其易产生m/z 206.0825的碎片离子峰,并通过脱羧产生m/z 178.087 1峰。两种化合物的质谱图见图3。以雷公藤晋碱为例,解析此类化合物的裂解规律,见图4。因此得出吡啶二羧酸部分含有羟基的生物碱会产生m/z 204系列的特征碎片离子峰,不含羟基的生物碱则产生m/z 206系列的特征碎片离子峰。
3.2.4 萜类化合物结构解析
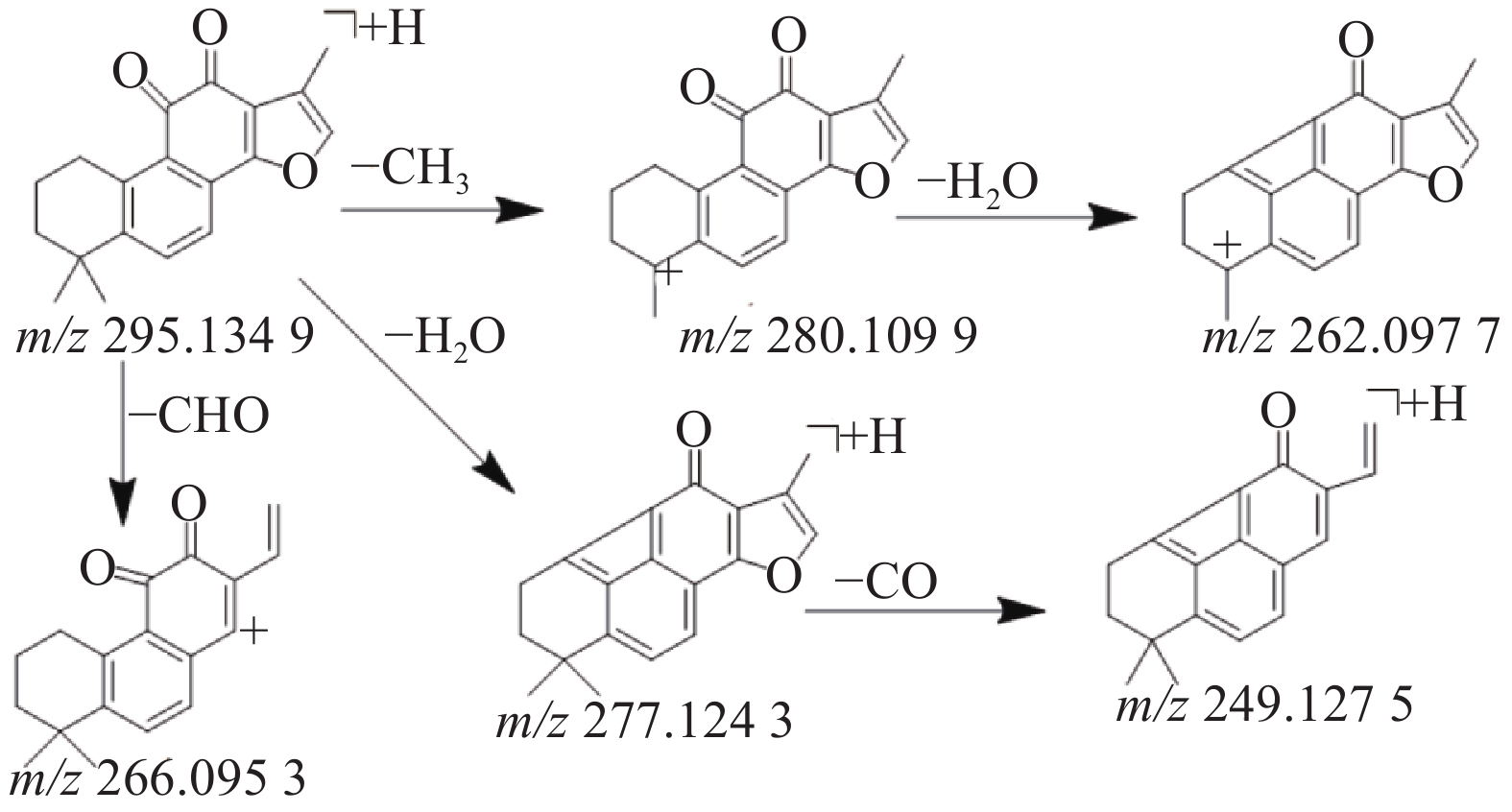
本研究共鉴定出8种萜类化合物,其中源于丹参药材中的5种萜类成分,丹参中的萜类化合物因其结构中主要含有羟基,羰基等取代基,所以质谱碰撞中主要丢失H2O,CO和-CH3等中性分子,产生一系列的碎片离子峰。化合物41在正离子模式下产生m/z 295.134 9的[M+H]+准分子离子峰,二级质谱中产生脱去1分子甲基形成的m/z 280.1099的碎片离子峰,在此基础上有丢失1分子水形成m/z 262.097 7峰,准分子离子峰脱去1分子H2O或脱去1个-CHO形成m/z 277.124 3峰或m/z 266.095 3峰,m/z 249.127 5峰是m/z 277.124 3脱去1分子H2O形成的碎片峰,通过对照品的保留时间和参考文献[10,18,23]数据比对,鉴定该化合物为丹参酮Ⅱ A,其质谱裂解规律见图5。
来源于雷公藤药材中的3种二萜类成分,该类化合物的二级质谱中出现一系列的脱水、脱CO和异丙基等碎片离子峰。化合物11在正离子模式下产生m/z 361.166 5的准分子离子峰,脱去2分子H2O和2分子CO形成m/z 269.154 3的碎片离子峰,m/z 227.108 3为m/z 269.154 3脱去1分子CH2CHCH3形成的碎片离子,其进一步脱1分子H2O和HCHO形成m/z 185.096 9的碎片离子,通过对照品比对和参考文献[14-15]的质谱数据,确定化合物10为雷公藤甲素。化合物18在正离子模式下产生m/z 359.148 9的准分子离子峰,脱去2分子H2O和2分子CO形成m/z 267.138 0的碎片离子峰,m/z 225.019 5为m/z 267.138 0脱去1分子CH2CHCH3形成的碎片离子,其进一步脱1分子H2O和HCHO形成m/z 183.0799的碎片离子,通过对照品比对和参考文献[19]的质谱数据,确定化合物16为雷公藤内酯酮。化合物39在负离子模式下产生m/z 299.199 6 的准分子离子峰,二级质谱中产生m/z 283.168 2的碎片离子, 提示为丢失1个-CH3后形成双键产生的碎片离子峰,A环发生RDA裂解产生m/z 213.090 8的碎片离子峰,通过数据库比对和参考文献[22]的质谱数据,推测化合物39为雷酚萜。
3.2.5 酚酸类化合物结构解析
在正负离子模式下共鉴定出乙酸乙酯部位中5种酚酸类成分,均来自于丹参药材,参考文献[18]报道的丹参中酚酸类成分的裂解规律发现,酚酸类化合物主要含有羰基、羧基和羟基,所以在质谱碰撞中易丢失CO、H2O和CO2的中性碎片;丹参素和咖啡酸作为基本母核而其他的水溶性酚酸类化合物大多数为这两者的聚合或缩合产物,主要为缩酚酸类的成分,在质谱碰撞中易丢失[M-H-180]−和[M-H-198]−中性碎片;含有羧基的单体化合物在负离子模式下会产生135[C8H7O2]−和179[C9H7O4]−的特征性碎片。化合物5中,在负离子模式下产生m/z 197.044 7的[M-H]−准分子离子峰,二级质谱进一步产生丢失1分子H2O和1分子CO2,形成的m/z 179.038 3和m/z 135.044 3的碎片离子峰,推测出结构中含有羧基,结合其精确分子量和参考文献[10]质谱数据,推测该化合物为丹参素。化合物12中,负离子模式下产生m/z 491.097 0的[M-H]−准分子离子峰,二级质谱中产生m/z 311.054 9和m/z 293.044 6的碎片离子峰,分别为[M-H-180]−和[M-H-198]−, m/z 267.064 6峰为m/z 311.054 9脱去1分子CO2所产生,根据m/z 135.044 7峰推测结构中含有羧基,结合其精确分子量和参考文献[18]质谱数据的比较,推测该化合物为丹酚酸C。化合物1中,正离子模式下给出m/z 139.039 4的[M+H]+准分子离子峰,脱去1分子H2O形成m/z 121.028 7的碎片离子峰,通过数据库比对和参考文献[10],推测该化合物1为原儿茶醛。化合物13中,在正离子模式下产生m/z 341.066 9的[M+H]+准分子离子峰,脱去1分子CO2形成m/z 295.060 7的碎片离子峰,m/z 277.050 9和m/z 249.056 0的碎片离子峰是m/z 295.060 7峰分别脱去1分子H2O和1分子CO2形成的,通过数据库比对和参考文献[17]质谱数据,推测该化合物为丹酚酸G。
4. 讨论
4.1 色谱与质谱条件考察
本实验流动相考察了乙腈-水系统和甲醇-水系统,结果乙腈-水系统中化合物的分离度较好,加入甲酸可以改善峰形,有助于化合物的离子化,提高质谱的响应,最终选择乙腈-0.1%甲酸水系统作为本次研究的流动相。
4.2 化学成分的定性分析
据以往文献中三色片各化学成分的研究报道,收集各药材的主要化学成分的精确分子量,碎片离子峰等信息,建立相应的化学成分数据库。通过数据库比对,对照品保留时间及参考文献中质谱数据鉴定三色片醇提物乙酸乙酯部位的化学成分。本研究共鉴定出42个化合物,其中5个是通过对照品鉴定得出,对无对照品的化合物,通过质谱的裂解特征及参考文献进行结构表征,对同分异构体应结合其在液相色谱中化合物的保留时间及质谱行为,综合对其定性鉴别。
三色片醇提物的乙酸乙酯部位具有较强的抗补体活性,本研究采用UPLC-Q-TOF-MS法对其中的化学成分进行结构表征,结果发现该部位主要含有生物碱类,萜类,黄酮和酚酸类等化学成分。其中以来源于雷公藤药材中极性中等的生物碱类成分含量较多,这与三色片提取物的制备工艺有关,三色片中雷公藤药材采用乙醇加热回流提取的方式,而黄芪和丹参药材采用水提取醇沉淀的方式。此外,先前的研究发现广藿香中的黄酮和萜类化合物对旁路途径的补体激活具有明显的抑制作用,紫花地丁中的生物碱类成分对旁路途径也有抑制作用(AP50=0.22~0.50 g/L), 牡丹皮和毛七公的抗补体活性成分研究中发现酚羟基决定抗补体活性的存在与否,没食子酰基可改善抗补体活性,甲基则对抗补体活性不利[25-27]。通过本次研究对三色片醇提物的乙酸乙酯部位的化学成分进行了初步表征,为阐明三色片的药效物质基础提供参考依据。研究的不足之处在于,仍有部分化学成分尚未定性鉴定,含量较高的单体成分未进行体外抗补体活性的测定,未来将通过中药化学的方法获得含量较高的单体成分,并进行结构鉴定和抗补体活性测定。
-
表 1 不同比活度11C-FMZ注射后在不同时间的百分注射剂量率(%ID/L)
测定成分 时间(t/min) 1 2 3 4 5 10 15 20 30 40 60 11C-FMZ① 9.76±4.80 6.00±1.42 3.30±0.60 2.77±0.39 2.25±0.33 1.95±0.24 1.55±0.15 1.28±0.18 1.09±0.14 0.98±0.15 0.78±0.10 羧酸代谢物① 0.04±0.04 0.19±0.10 0.44±0.13 0.51±0.14 0.67±0.13 1.88±0.24 2.00±0.33 1.73±0.28 1.50±0.28 1.59±0.20 1.22±0.22 11C-FMZ② 13.84±4.67 6.10±1.31 3.27±0.49 3.10±0.36 2.66±0.37 1.97±0.33 1.42±0.13 1.23±0.15 1.11±0.06 0.67±0.13 0.45±0.07 羧酸代谢物② 0.15±0.11 0.28±0.08 0.50±0.17 0.55±0.29 0.98±0.27 2.22±0.42 2.36±0.44 2.39±0.34 1.90±0.41 1.39±0.25 0.98±0.14 注:①表示高比活度(268.3±57.2)×103 Ci/mol注射后的剂量率;②表示低比活度(57.8±11.4)×103Ci/mol注射后的剂量率。  下载: 导出CSV
下载: 导出CSV
表 2 不同比活度11C-FMZ的羧酸代谢物在不同时间百分注射剂量率的占比(%)
11C-FMZ羧酸代谢物 时间(t/min) 1 2 3 4 5 10 15 20 30 40 60 高比活度 0.54±0.54 3.32±1.89 12.04±3.61 15.58±4.25 23.11±4.92 48.98±4.89 56.11±4.68 57.36±5.63 57.60±3.45 61.92±4.51 60.85±5.39 低比活度 1.12±0.70 4.53±1.69 13.48±4.94 14.64±6.01 27.04±7.81 52.90±5.81 62.04±4.69* 65.85±5.35* 62.54±5.75* 67.26±4.88* 68.26±4.47* *P<0.05,与高比活度组比较。
下载: 导出CSV
-
[1] 侯成, 卢光照, 张翮, 等. 氟马西尼不同给药途径的催醒作用评价[J]. 药学实践杂志, 2018, 36(1):30-33, 54. doi: 10.3969/j.issn.1006-0111.2018.01.006 [2] 李奇明, 金榕兵, 王方洋, 等. PET示踪剂11C-氟马西尼的合成及质量控制[J]. 同位素, 2017, 30(1):16-22. doi: 10.7538/tws.2017.30.01.0016 [3] PARENTE A, VÁLLEZ GARCÍA D, SHOJI A, et al. Contribution of neuroinflammation to changes in[11C]flumazenil binding in the rat brain: Evaluation of the inflamed Pons as reference tissue[J]. Nucl Med Biol,2017,49:50-56. doi: 10.1016/j.nucmedbio.2017.03.001 [4] 张锦明, 田嘉禾, 王武尚, 等. 在线制备11C-Triflate-CH3[J]. 同位素, 2006, 19(2):124-128. doi: 10.3969/j.issn.1000-7512.2006.02.014 [5] 石庆学, 左峰, 郭佳, 等. GABAa/BZ受体示踪药物碳11-氟马西尼的全自动合成[J]. 中国药师, 2018, 21(12):2232-2234. doi: 10.3969/j.issn.1008-049X.2018.12.038 [6] 赵惠扬. 核医学[M]. 上海: 上海科学技术出版社, 1981. [7] ISHIWATA K, ITOU T, OHYAMA M, et al. Metabolite analysis of [11C] flumazenil in human plasma: assessment as the standardized value for quantitative PET studies[J]. Ann Nucl Med,1998,12(1):55-59. doi: 10.1007/BF03165418 [8] DEBRUYNE D, ABADIE P, BARRE L, et al. Plasma pharmacokinetics and metabolism of the benzodiazepine antagonist [11C] Ro 15-1788(flumazenil) in baboon and human during positron emission tomography studies[J]. Eur J Drug Metab Pharmacokinet,1991,16(2):141-152. doi: 10.1007/BF03189951 [9] KLOTZ U, ZIEGLER G, REIMANN I W. Pharmacokinetics of the selective benzodiazepine antagonist Ro 15-1788 in man[J]. Eur J Clin Pharmacol,1984,27(1):115-117. doi: 10.1007/BF02395217 [10] SAMSON Y, PAPPATA S, HANTRAYE P, et al. Recepteurs centraux aux benzodiazepines: etudes chez l’homme en tomographie d'emission de positons[J]. Circul Metabol Cerv,1987,4:155-166. [11] SHINOTOH H, YAMASAKI T, INOUE O, et al. Visualization of specific binding sites of benzodiazepine in human brain[J]. J Nucl Med,1986,27(10):1593-1599. [12] PAPPATA S, SAMSON Y, CHAVOIX C, et al. Regional specific binding of [11C] RO 151788 to central type benzodiazepine receptors in human brain: quantitative evaluation by PET[J]. J Cereb Blood Flow Metab,1988,8(3):304-313. doi: 10.1038/jcbfm.1988.65 [13] PERSSON A, PAULI S, HALLDIN C, et al. Saturation analysis of specific 11C Ro 15-1788 binding to the human neocortex using positron emission tomography[J]. Hum Psychopharmacol Clin Exp,1989,4(1):21-31. doi: 10.1002/hup.470040105 [14] KLUMPERS U M, VELTMAN D J, BOELLAARD R, et al. Comparison of plasma input and reference tissue models for analysing [11C] flumazenil studies[J]. J Cereb Blood Flow Metab,2008,28(3):579-587. doi: 10.1038/sj.jcbfm.9600554 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 6515
- HTML全文浏览量: 2258
- PDF下载量: 16
- 被引次数: 0




