

 下载:
下载:


 下载:
下载:

-
信号转导和转录激活因子3(signal transducer and activator of transcription 3,STAT3)是一种信号转录蛋白[1]。该蛋白在结构上主要有N末端、卷曲螺旋、DNA接合、连结、SH2和转录激活区域6个位置域[2]。现有的研究结果表明,STAT3抑制剂可靶向作用于STAT3蛋白的不同结构区域,抑制其在细胞中的表达,为抗炎、抗癌治疗提供思路[3-4]。
-
近年来研究者对STAT3蛋白的探索不断加深,对其在体内的活化机制也有清晰的发现[5]。STAT3在细胞质中分析信号、转导,在细胞核中转录激活[6]。STAT3蛋白在细胞中扮演着重要角色,可影响细胞的生长凋亡机制,研究者们观察到失控的STAT3在异常激活中会导致细胞发生恶性转化,对肿瘤细胞的形成和发展具有重要影响[7]。
STAT3的激活可通过各种细胞因子、生长因子和激素与细胞表面的受体结合而实现。在Janus激酶(JAK)-STAT3信号途径中,细胞因子与细胞膜上的蛋白受体结合,成为二聚体。二聚体在细胞质中募集并激活JAK激酶蛋白,激活的JAK蛋白磷酸化受体酪氨酸残基。随后,STAT3的Tyr705残基被JAKs磷酸化,STAT3蛋白被活化再形成二聚体后,从细胞质转移到细胞核中,与特异性DNA反应元件结合,最终调节STAT3靶基因的表达[8]。
STAT3的每个结构域各有其独特的生物作用。STAT3的N末端结构域对STAT3二聚体核易位以及DNA结合具有重要作用;卷曲螺旋结构域对STAT3与各种蛋白质的协同效应具有一定影响。DNA结构域可匹配特殊的DNA序列,然后形成STAT3-DNA复合物。连接区域参加与细胞转录系统的相互作用。SH2结构域对于STAT3二聚化也至关重要。因为在肿瘤细胞中的过度激活现象,以及抗STAT3的抑制剂对于抗炎免疫具有良好效果,STAT3已成为近年来药物研发的一个热门方向,研究人员纷纷投入巨大热情,不断挖掘STAT3的医药价值[9-10]。
-
STAT3抑制剂可靶向作用于STAT3蛋白,抑制其在癌细胞中的磷酸化、二聚化以及核易位,使其转录功能丧失,有可能为疾病治疗提供新方向[11]。
-
Turkson等[12]研究了STAT3的结合肽PY*LKTK(Y*代表磷酸酪氨酸)在体外破坏STAT3活性的能力。核提取物中PY*LKTK的存在会导致STAT3的DNA结合活性水平显著降低,因此破坏了核易位。作为肽直接抑制STAT3的功能重要性的证据,发现PY*LKTK-mts(mts,膜移位序列)可在体内选择性抑制组成型和配体诱导的STAT3激活。此外,PY*LKTK-mts可抑制Src癌蛋白的转化。Turkson通过确定了抑制STAT3信号转导的最小肽,为使用该肽作为新型拟肽药物进行临床治疗STAT3异常激活的疾病提供了概念基础。
Timofeeva等[13]在表征STAT3-N末端域的选择性抑制剂ST3-H2A2的作用中,观察到该化合物使凋亡基因强烈激活,在癌细胞中诱导细胞凋亡。通过DNA-chip技术和平铺人类启动子阵列技术,发现响应ST3-H2A2的基因在表达激活的同时会伴随着STAT3染色质结合而改变。此外,将siRNA敲低可证实ST3-HA2A对基因表达和染色质结合的影响是STAT3依赖性的,而C/EBP同源蛋白(CHOP)启动子的STAT3结合区位于癌细胞中染色质的DNase I超敏位点中,而不是未转化细胞中,这表明STAT3结合和抑制作用极有可能与染色质结构有关。因此我们猜想,STAT3-N末端选择性抑制剂可调节癌细胞凋亡基因的表达,促使细胞凋亡,为癌症治疗提供新方案。
-
Huang等[14]使用计算机虚拟筛选(AlloFinder)的途径对STAT3的C端卷曲螺旋区域(CCD)进行匹配,通过对分数排名前15的化合物进行生物测定,筛得理论最佳化合物K116(图1)。在此化合物的作用下,荧光偏振显示STAT3的IC50值为7.99 μmol/L。在免疫印迹实验中,STAT3的Y705磷酸化被抑制,而上游的激酶Src及其磷酸化无影响。同时,在肿瘤细胞株MDAMB-468及DU145中,K116表现出对STAT3很强的抑制,IC50为4.46和23.85 μmol/L,表明K116具有良好的特异性抑制STAT3的作用。从K116的研究历程来看,未来进行虚拟筛选的计算化学方法将逐步成为药物研发的一种趋势,在此趋势发展的初期,我们还需要不断改进底层算法并完善数据库内容。
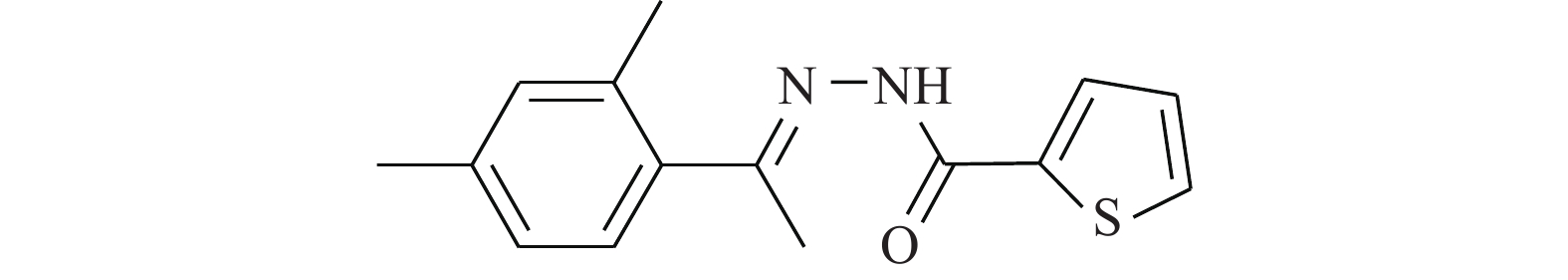
图 1 化合物K116的化学结构
-
Huang等[15]通过使用改进的虚拟筛选策略靶向STAT3的DNA结合域(DBD),得到了靶向STAT3-DBD的小分子化合物inS3-54(图2)。采用[32P]标记的DNA双链探针及电泳迁移率变动分析表明,inS3-54选择性抑制STAT3与DNA的结合,而对STAT1无影响。与非癌细胞相比,inS3-54优先抑制癌症细胞的增殖,抑制STAT3下游靶基因的表达和STAT3原位结合染色质。因为inS3-54不与SH2结构域相连接,不抑制STAT3二聚化,但可以抑制STAT3与基因组DNA的结合。inS3-54代表了一种新型探针,对于开发靶向STAT3的DBD域特异性抑制剂具有积极意义。
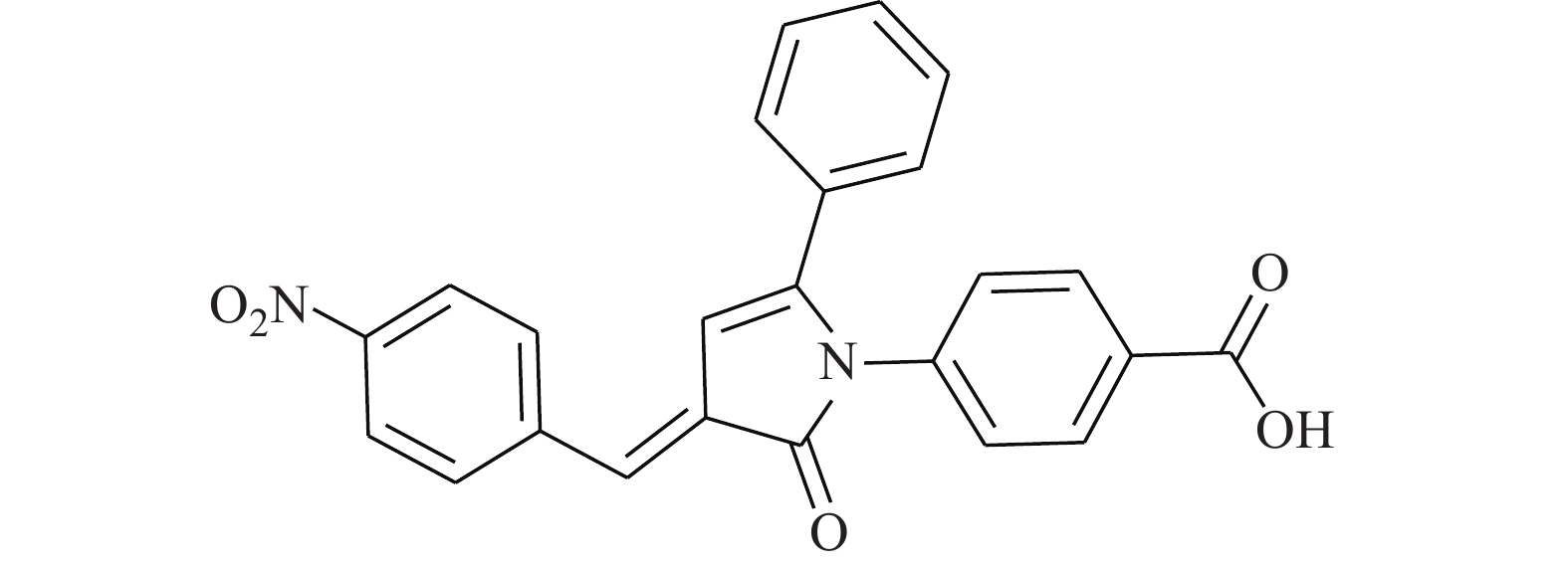
图 2 化合物inS3-54的化学结构
Buettner等[16]为了证明化合物C48(图3)可作为STAT3的选择性抑制剂,使用定点诱变和多种生化技术,探明C48将STAT3中的Cys468烷基化的过程。进一步研究证明,C48会在STAT3过度表达的肿瘤细胞系中阻止STAT3核易位,导致小鼠体内肿瘤生长的显著抑制。这些发现表明,STAT3中的Cys468代表了一个新的治疗干预位点,并推测烷基化有望成为STAT3相关类型癌症的潜在治疗方法。

图 3 化合物C48的化学结构
-
Cheng等[17]研究化合物6-OAP(图4)与肺癌细胞的作用关系,发现该化合物在H1975和A549细胞中以剂量和时间依赖性的方式抑制了STAT3的转录(抑制位点Tyr705),进而抑制由IL-6等细胞因子诱导的STAT3的活化、磷酸化与二聚体的形成。6-OAP在STAT3的SH2结构域与Ser611/Ser613/Arg609形成氢键,阻碍IL-6诱导的STAT3磷酸化,对肺癌细胞和对S期激酶相关蛋白转录有抑制作用。药理实验表明,6-OAP抑制静脉注射肺癌细胞的SCID小鼠的肿瘤生长。6-OAP的效果优异,毒副作用较小,有望进一步挖掘其药用价值。
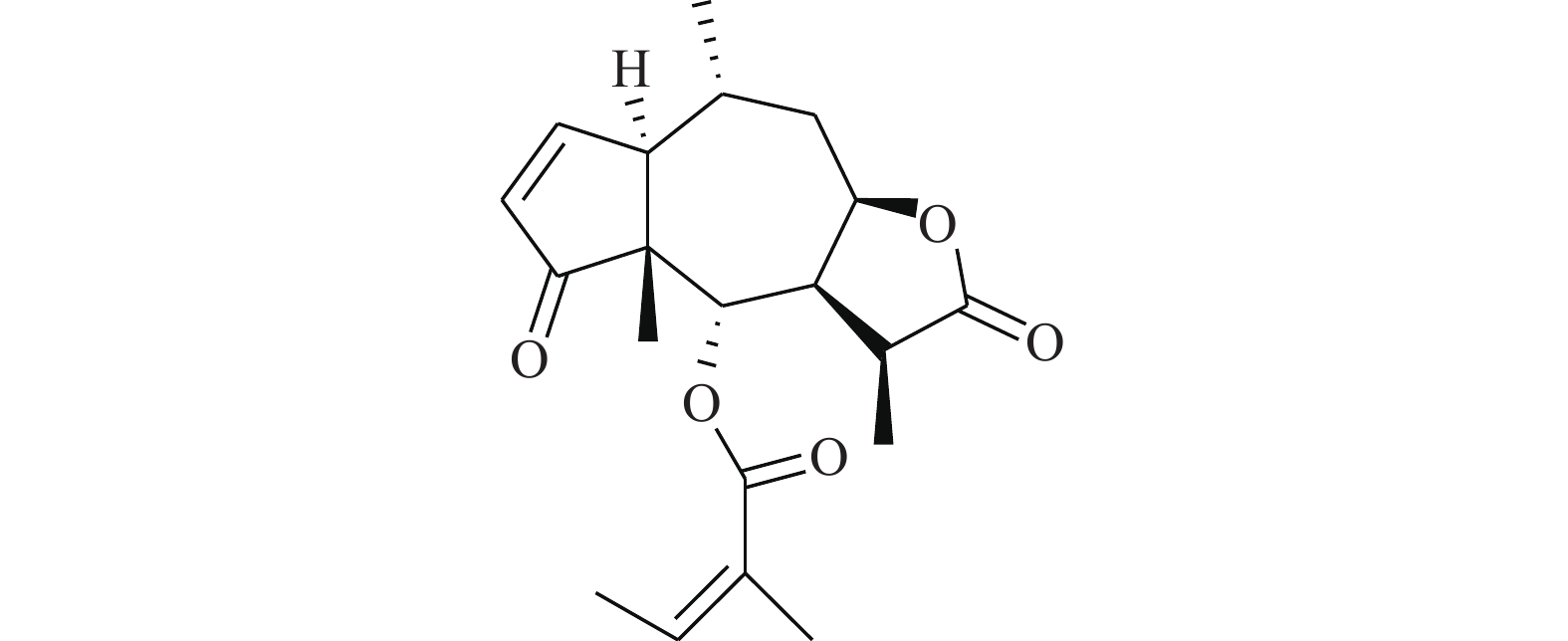
图 4 化合物6-OAP的化学结构
Schust等[18]从化学数据库中通过模拟筛选得到化合物Stattic(图5)。Stattic能有效抑制STAT3的SH2结构域,对STAT3的激活、二聚化和核易位等关键步骤有阻滞作用。将MDA-MB-231和MDA-MB-435S细胞在指定浓度下用Stattic处理2 h,对STAT3、p-STAT3和其他信号分子(JAK1/JAK2等)的全细胞裂解物进行蛋白质印迹分析,证明了Stattic对STAT3的磷酸化有显著的抑制效应,且其特异选择性较高,有潜力成为高效的STAT3抑制剂。
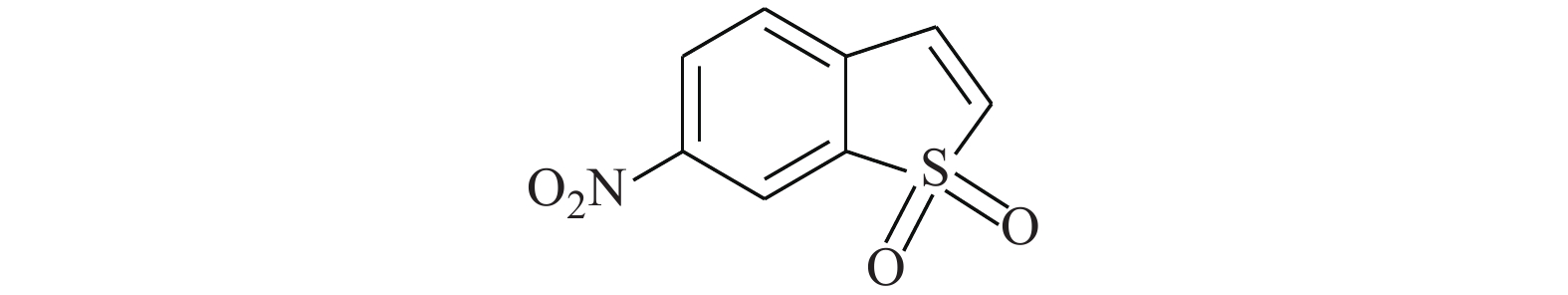
图 5 化合物Stattic的化学结构
Chen等[19]通过传统的有机合成方法得到了化合物HJC0152(图6),发现其对人乳腺癌和胰腺癌细胞显示出比氯硝柳胺相似或更高的效力。经过结构修饰后(将苯环上的烷氧基改为氨基)的HJC0152显示出优异的水溶性,与氯硝柳胺相比提高了约3 300倍。在光学显微镜的观察下,将MDA-MB-231乳腺癌细胞用化合物HJC0152处理48 h,观察细胞形态变化,发现细胞周期进程被抑制,细胞凋亡变得更加常见。在带有乳腺肿瘤异种移植物的裸鼠中,HJC0152显著抑制了肿瘤的生长。从这些结果中推测,具有更好水溶性的化合物HJC0152有望被开发成用于癌症治疗的口服生物利用剂。
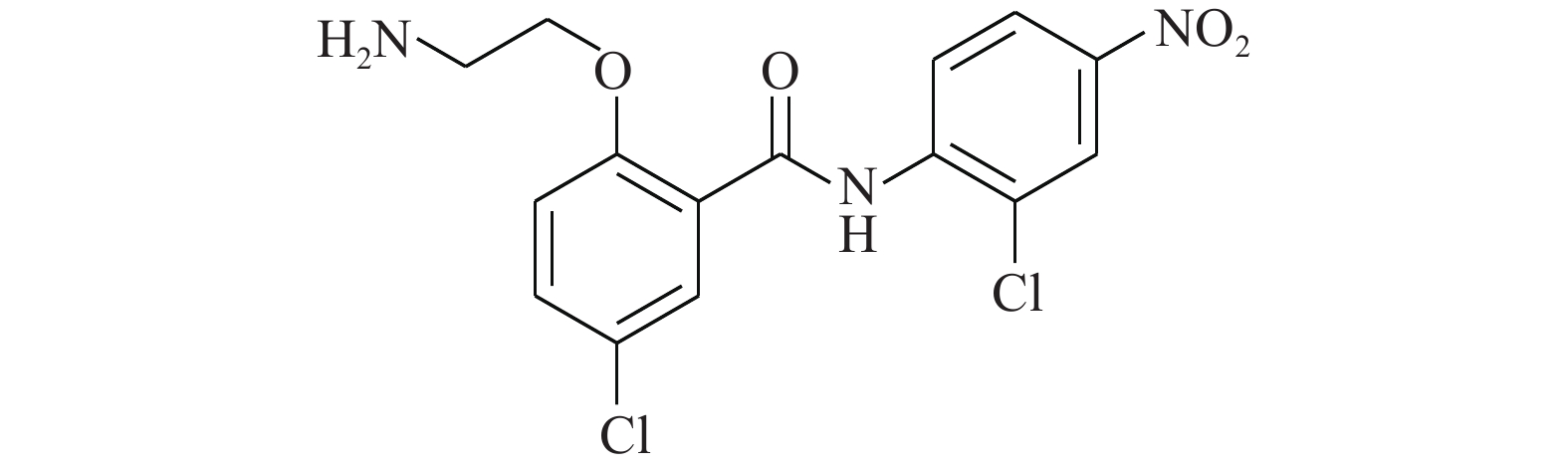
图 6 化合物HJC0152的化学结构
Shin等[20]首次报道了隐丹参酮(图7)抗癌活性是通过抑制STAT3实现的。隐丹参酮可快速抑制DU145人前列腺癌细胞中的STAT3的Tyr705磷酸化,降低STAT3下游靶蛋白的表达。隐丹参酮抑制STAT3磷酸化是由独立于JAK2的机制引起的,且不影响上游酪氨酸激酶。隐丹参酮可直接与STAT3分子结合(STAT3的SH2结构域),抑制其二聚化,并下调细胞周期蛋白D1、Bcl-xL和survivin的表达,使G0-G1期的细胞蓄积,降低STAT3转录调控活性。这些结果表明,隐丹参酮是良好的STAT3靶向抑制剂。
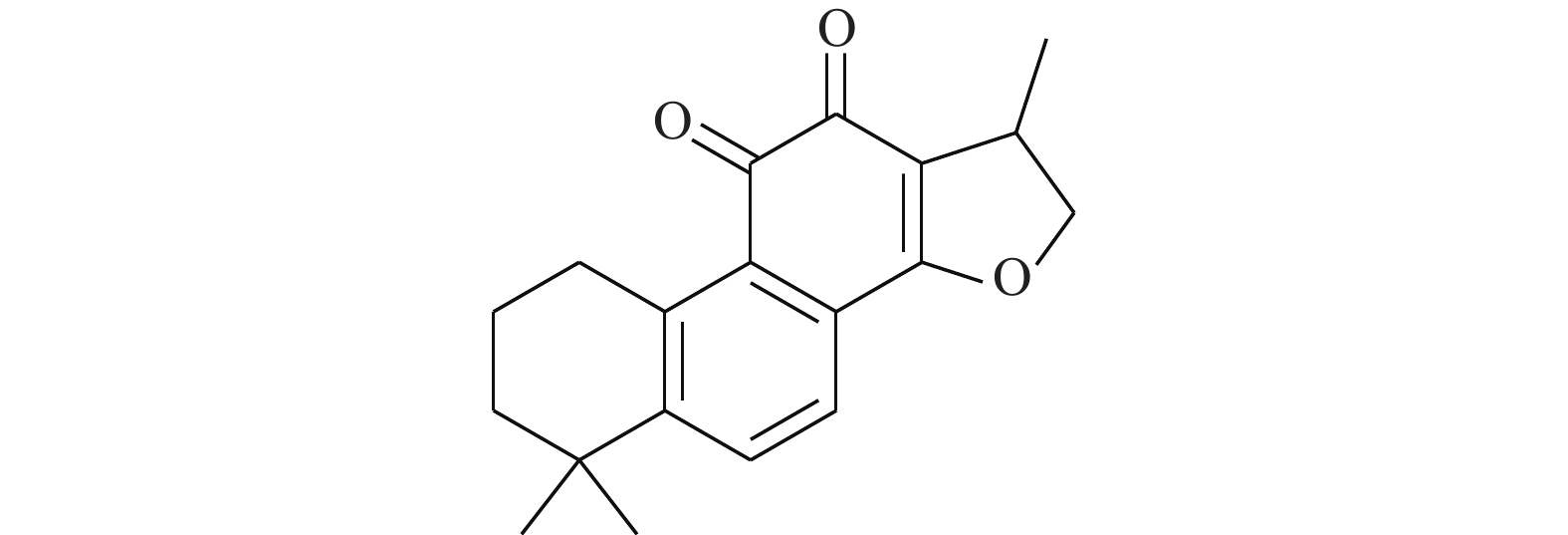
图 7 隐丹参酮的化学结构
Iwamoto等[21]研究发现,苯达莫司汀(BENDA,图8)作为一种烷化剂,对多种癌症具有临床活性,包括但不限于非霍奇金淋巴瘤、慢性淋巴细胞性白血病和多发性骨髓瘤等。BENDA可以选择性结合细胞中的STAT3并抑制其活性,BENDA在体外选择性地拮抗STAT3-SH2结构域(IC50=7.4 μmol/L),而不是其他含有SH2的蛋白质(拮抗STAT1-SH2的IC50为60 μmol/L)。BENDA结合到STAT3中的半胱氨酸残基Cys550和Cys712位置上,从而抑制SH2与相应的p-Tyr肽的结合。
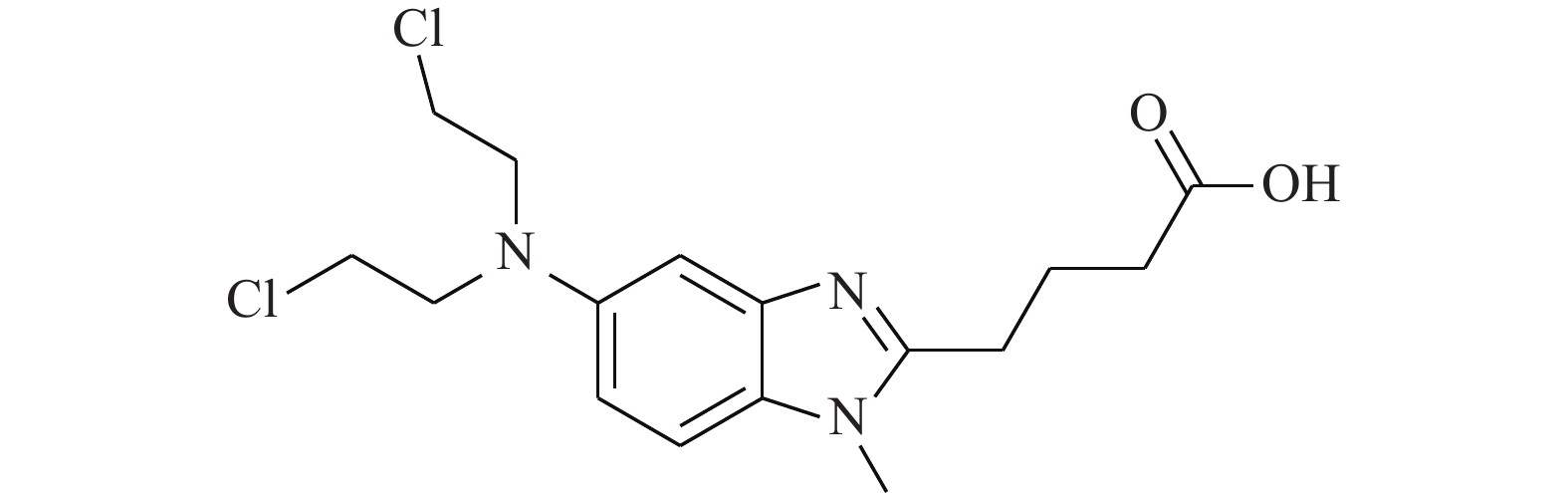
图 8 苯达莫司汀的化学结构
Chang等[22]研究发现,STAT3抑制剂BBI608(图9)是降低EGFR阳性肺癌细胞系细胞活力的潜在药物。BBI608可降低组蛋白甲基转移酶G9a介导的表皮生长因子受体3表达,抑制EGFR阳性肺癌(包括EGFR E746-A750 HCC827、WT-A549和T790 M H1975)的细胞活力。在与阿法替尼的对照实验中,BBI608更为明显地降低了A549和H1975细胞的生存能力,并降低了A549的细胞迁移。因此,BBI608极有潜力成为EGFR阳性肺癌的治疗药物。
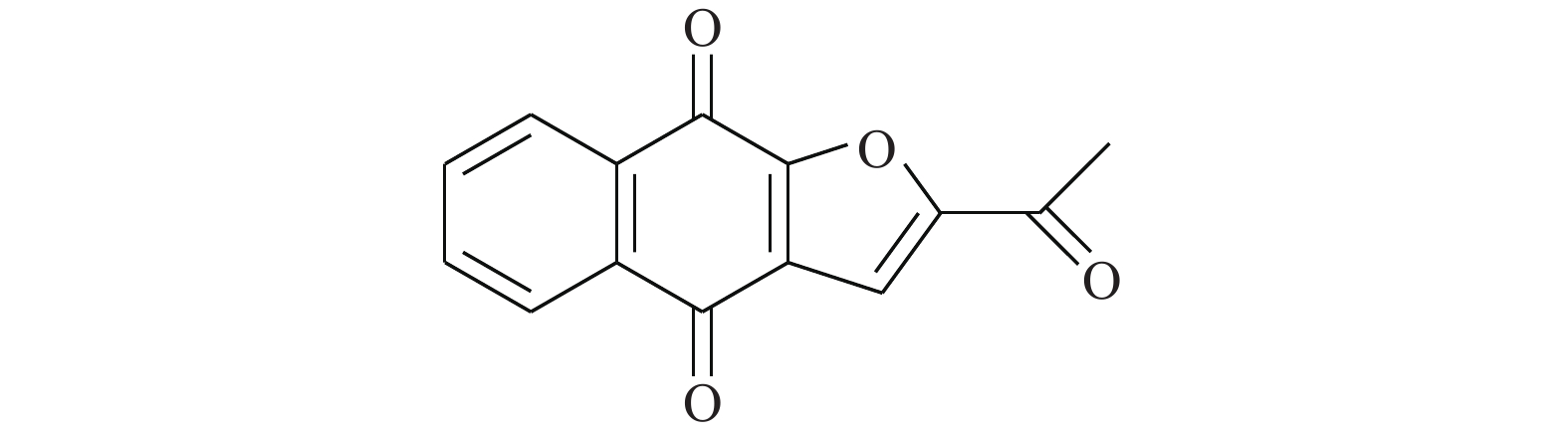
图 9 化合物BBI608的化学结构
研究表明,STAT3在各种类型的癌症中被组成性激活,并且直接参与癌细胞的免疫调节。Shastri等[23]对骨髓增生异常综合征(MDS)和急性髓性白血病(AML)患者的造血干细胞和祖细胞群体进行转录组分析,发现STAT3存在异常激活现象,而敲除STAT3后,体内白血病细胞的生长也受到了抑制,各类癌基因在恶性细胞中的表达也随之下降。
-
STAT3在多种实体瘤和血液性肿瘤中会过度激活,对肿瘤的发生和发展有着重要影响。因此,STAT3作为治疗肿瘤的新靶点已被研究人员所认同;同时,STAT3抑制剂对抗炎免疫有重要作用,在类风湿性关节炎等疾病研究中颇有成效。21世纪以来,STAT3抑制剂的临床前研究不断深入,但进入临床使用的药物却很少。因此,研发高效、低毒的STAT3抑制剂仍将是一种挑战。近年来,研究人员将多种天然产物(如雷公藤红素等)作为先导化合物,合成了更有效的小分子化合物,通过不断筛选、评价和选择活性大、特异性强的STAT3抑制剂,将中西医药物治疗与放疗、化疗结合,这可能是今后的STAT3研究方向之一,为抗肿瘤以及抗炎免疫研究提供新方向。
Research progress of STAT3 inhibitors
-
摘要: 信号转导及转录激活因子3(signal transducer and activator of transcription 3,STAT3)是一种信号转录蛋白。STAT3的异常激活与细胞的增殖、分化、癌变等密切相关,其在乳腺癌、胰腺癌、淋巴癌、肺癌等癌干细胞中存在异常表达。因此,抑制STAT3的异常表达已成为抗炎免疫、抗肿瘤治疗的一种新思路。Abstract: Signal transducer and activator of transcription 3 (STAT3) is a signal transcription protein that exists in the cytoplasm. The abnormal activation of STAT3 is closely related to cell proliferation, differentiation, and canceration. It has abnormal expression in cancer stem cells such as breast cancer, pancreatic cancer, lymphoma, and lung cancer. Therefore, inhibiting the abnormal expression of STAT3 has become a new approach for antitumor therapy.
-
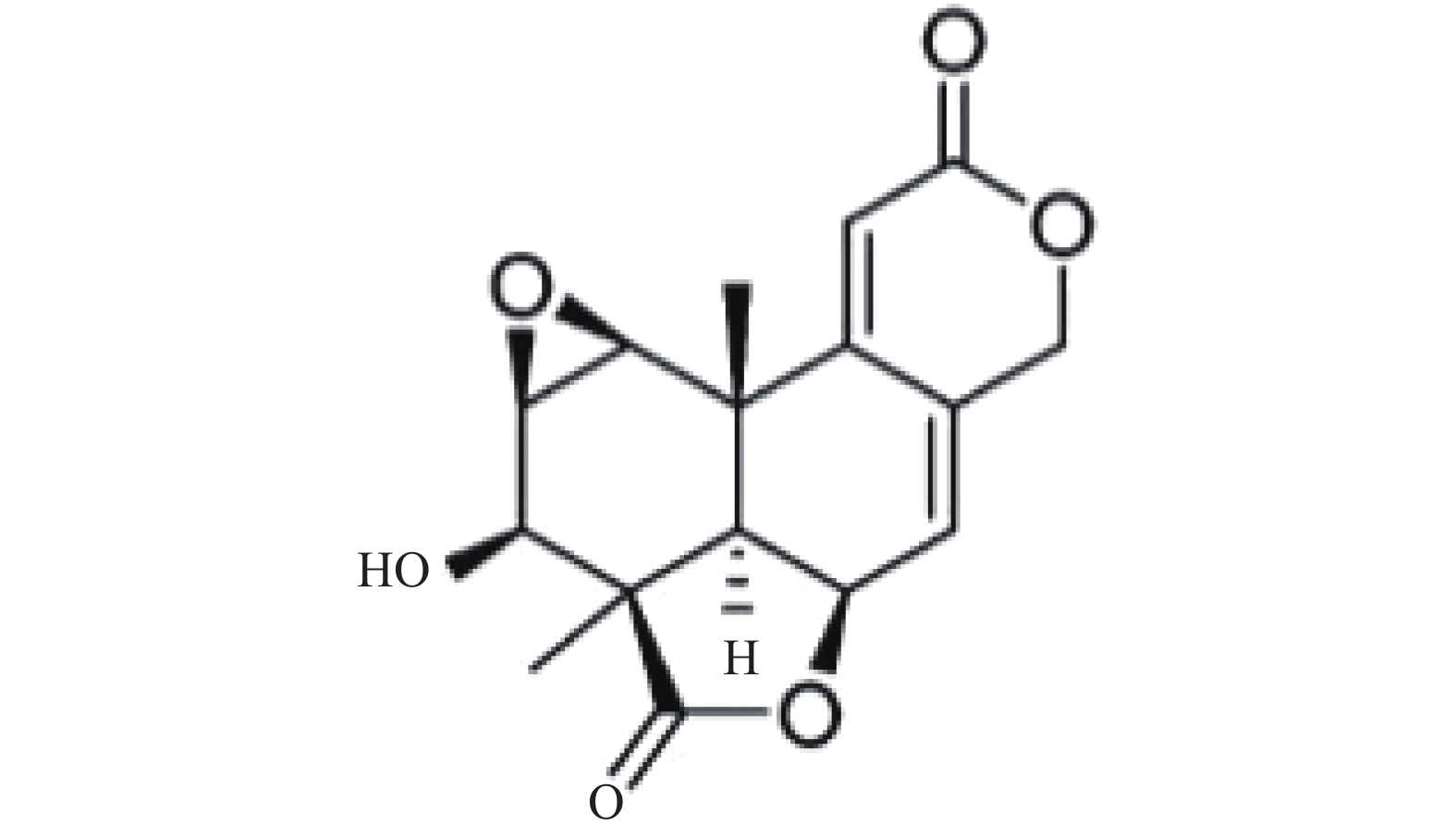
Wentilactone A(WA)是从海洋微生物中分离得到的去甲二萜类小分子化合物,对小细胞肺癌细胞系NCI-H460和NCI-H446细胞的增殖具有抑制作用,可诱导小细胞肺癌细胞系NCI-H446和NCT-H1688细胞凋亡[1]。对小细胞肺癌细胞系NCT-H1688细胞的细胞迁移和集落形成具有抑制作用[2]。WA作为一种有效抗肿瘤候选物,已从制备工艺、理化性质、急性毒性、生殖毒性等方面证明其良好的成药性[3]。遗传毒性安全性实验更是对其临床前研究全面性的补充,为提高临床用药的安全性,为了验证其是否具有遗传毒性,本实验应用微生物回复突变试验(Ames试验)、体外培养CHO细胞染色体畸变试验和小鼠骨髓微核试验方法对WA的遗传毒性进行了系统的研究,为WA的临床前毒性评价提供资料[4]。
1. 材料与方法
1.1 材料
1.1.1 化合物WA
Ames试验用WA(含量:99.9%,批号:20121115,黄色粉末)、体外培养CHO细胞染色体畸变试验用(规格:10 ml,含量:50.8 mg/ml,批号:20130506,黄色澄明溶液)、小鼠骨髓微核试验用WA(规格:10 ml/支:100mg,含量:10.2 mg/ml,批号:20130407,黄色澄明溶液),均由海军军医大学基础医学院生物化学与分子生物学教研室提供。WA的化学结构见图1。
1.1.2 菌株
组氨酸缺陷型鼠伤寒沙门菌(S. typhimurium)TA97、TA98、TA100、TA102和TA1535共5支菌株,复旦大学公共卫生学院环境卫生教研室赠予。实验前对其进行鉴定(R因子和自发回变数鉴定),均符合规定标准。
1.1.3 细胞
中国仓鼠卵巢(CHO)细胞由复旦大学公共卫生学院毒理教研室赠予。
1.1.4 动物
ICR小鼠(SPF级)共60只,每组10只,雌雄各半,5~6周龄,购入时体重16.5~20.8 g,由上海西普尔-必凯实验动物有限公司提供,动物质量合格证号:2008001629237。自购入起3天进行检疫,给药时体重20.3~23.6 g。
1.2 试验方法
按《新药(西药)临床前研究指导原则汇编》[5-6]的设计要求,分别应用Ames试验[7]、体外培养CHO细胞染色体畸变试验[8]和小鼠骨髓微核试验[9]检测WA的遗传毒性。检测终点覆盖了基因突变、染色体畸变和细胞有丝分裂异常。
1.3 实验步骤
1.3.1 Ames实验
根据《药物遗传毒性研究技术指导原则》的要求,应用S. typhimurium TA97、TA98、TA100、TA102和TA1535共5支菌株,设每皿5 000、500、50、5、0.5 μg 5个剂量组。此外,设空白对照、溶剂对照和阳性对照组(具体剂量见表1)。采用标准平板掺入法,使细菌在加和不加代谢活化系统S9的条件下接触受试物,每皿均加入供试品或溶剂对照DMSO 0.1 ml,每个剂量组及对照组均设3个平行皿。并用最低极限的琼脂培养基培养48 h后,先用显微镜观察平皿上的菌苔生长情况,确定受试物无明显的抑菌或杀菌作用,再人工计数每皿回复突变的菌落数,记录原始数据,并计算每组的均值和标准差,与溶剂对照组进行比较,重复实验一次。
表 1 阳性对照品名称及浓度组别 菌株 阳性对照品 溶液终浓度(μg/皿) 加入量(μl/皿) 浓度(μg/ml) -S9组 TA97 敌克松 50 100 500 TA98 敌克松 50 100 500 TA100 甲基磺酸甲酯 1 100 10 TA102 甲基磺酸甲酯 1 100 10 TA1535 4-硝基喹啉-N-氧化物 0.5 100 5 +S9组 TA97 2-氨基芴 10 100 100 TA98 2-氨基芴 10 100 100 TA100 2-氨基芴 10 100 100 TA102 1,8-二羟基蒽醌 50 100 500 TA1535 环磷酰胺 50 100 500 1.3.2 染色体畸变试验
在加和不加代谢活化系统S9的条件下,体外培养的CHO细胞中加入相应浓度的供试品或对照品,反应体系总体积为10 ml。低、中、高剂量组供试品终浓度分别为23.74、47.48和94.96 μg/ml,阳性对照组丝裂霉素C和环磷酰胺的终浓度分别为0.5 μg/ml、60 μg/ml,另设溶剂对照组分别作用于细胞4 h后换液继续培养至24 h,和药物作用细胞24 h后收集细胞。收获细胞前4 h,加入终浓度0.2 μg/ml秋水仙素,培养结束收集细胞,经离心、低渗处理、固定、离心、制片和Giemsa染色,每个剂量组制备2~3张玻片标本。
镜检时每组观察200个染色体中分散良好、数目完整的中期分裂相细胞(若观察到大量染色体畸变细胞,如阳性对照组,分析细胞数可相应减少为至少100个细胞)。计数染色体或染色单体的断裂、缺失及其他类型结构异常的数目(裂隙和核内复制一般不作为畸变类型),记录原始数据计算畸变率。
1.3.3 小鼠骨髓微核实验
检疫期结束后分别按低、中、高组小鼠给药,分别予100、200、400 mg/kg剂量给药,其中高剂量组设两组分别在药物作用24 h和48 h后处死,并设阳性对照组和溶剂对照组。分别在药物作用24 h和48 h后处死小鼠,取股骨骨髓制成骨髓涂片,每只动物制2张涂片,经甲醇固定后用pH6.8的Giemsa染液染色。
每只动物镜检约2000个骨髓嗜多染红细胞(PCE),计数含微核的PCE数(MNPCE),计算微核发生率,同时记录200个PCE计数过程中观察到的正染红细胞(NCE)的数目,并计算PCE/NCE值。
1.4 剂量设计
对于易溶无毒的化合物,细菌实验最高浓度应达到5 mg/皿[10]。Ames试验设每皿5 000、500、50、5、0.5 μg 5个剂量组,每皿均加入相应浓度的供试品溶液0.1 ml,另设空白对照、溶剂对照和阳性对照。在哺乳动物细胞体外遗传实验中,毒性水平应高于50%细胞抑制率或细胞融合率[11]。通过预实验确定供试品的IC50为94.96 μg/ml。染色体畸变试验的低、中、高剂量组为23.74、47.48和94.96 μg/ml,试验时在10 ml的试验体系中分别加入0.1 ml各剂量组的应用液,另设阳性对照组和溶剂对照。ICR小鼠的微核试验采用小鼠经尾静脉注射给药,总剂量分别为100、200、400 mg/kg(单次给药剂量分别为50、100、200 mg/kg,分上、下午两次经尾静脉注射给药),给药容积为20 ml/kg体重;同时设阳性对照组和溶剂对照组。阳性对照组以40 mg/kg体重的剂量腹腔注射环磷酰胺,给药容量为10 ml/kg体重;溶剂对照组给药方式与受试物组相同,以20 ml/kg体重的容积经尾静脉注射生理盐水。
1.5 统计方法
Ames试验结果的评价是以溶剂对照组的回复突变菌落数为基础,与受试物各剂量组相比较。若某剂量组回复突变菌落数为溶剂对照组的2倍以上,呈现可重复性,并在一定的剂量范围内存在着剂量-反应关系,则判断为阳性[12]。染色体畸变试验和小鼠骨髓微核试验均采用卡方检验或方差分析方法研究给药组与对照组之间是否具有统计学意义[13]。
2. 结果与分析
2.1 Ames试验结果
受试物各剂量组和对照组的平皿均可见背景菌苔生长。5支菌株的自发回复突变菌落数以及阳性对照品诱发的回复突变菌落数均在历史参考范围内,并且各菌株阳性对照组的回复突变菌落与空白对照组相比数目显著增加,提示本试验系统符合要求。在最高剂量已达到5 000 μg/皿的受试条件下,未观察到受试物的抑菌现象。各剂量组受试物在加或不加S9时对TA97、TA98、TA100、TA102和TA1535所诱发的回复突变菌落数均与自发的突变菌落数相近,未观察到明显的剂量-反应关系。结果见表2和表3。
表 2 WA对5支菌株的回变菌落数试验结果(个/皿,${\rm{\bar x}}$ ±s)(第1次)组别(μg/皿) TA97 TA98 TA100 TA102 TA1535 +S9 -S9 +S9 -S9 +S9 -S9 +S9 -S9 +S9 -S9 5 000 36±5 40±4 21±3 21±2 131±26 93±17 177±21 185±25 25±3 14±2 500 36±5 39±8 17±4 20±3 114±22 90±23 195±8 207±7 23±1 19±1 50 39±10 35±5 21±7 21±3 115±21 97±8 196±33 179±18 19±8 16±4 5 41±6 40±3 21±5 22±4 114±15 87±19 195±21 193±8 19±2 14±1 0.5 37±6 39±3 20±3 19±3 128±15 91±12 201±14 205±14 20±6 16±4 空白对照组 40±7 36±5 17±2 19±4 99±17 89±9 213±30 197±34 21±7 19±4 溶剂对照组 40±6 34±1 22±5 26±2 95±8 89±9 193±8 199±29 23±3 14±1 阳性对照组 839±24 841±47 936±56 1 077±55 968±27 1 111±66 1 101±17 1 024±53 342±44 345±13 表 3 WA对5支菌株的回变菌落数试验结果(个/皿,${\rm{\bar x}}$ ±s)(第2次)组别(μg/皿) TA97 TA98 TA100 TA102 TA1535 +S9 -S9 +S9 -S9 +S9 -S9 +S9 -S9 +S9 -S9 5 000 38±5 34±5 20±5 21±5 128±24 105±26 179±21 189±19 20±4 20±3 500 39±7 34±3 21±2 20±7 108±13 92±28 192±16 187±30 16±3 19±4 50 38±9 30±3 19±3 18±1 117±14 102±5 197±5 196±22 17±3 18±1 5 37±8 29±4 18±2 17±1 111±5 98±33 206±24 191±21 16±1 20±2 0.5 35±6 33±8 24±2 23±3 107±7 97±8 203±6 193±10 15±4 15±4 空白对照组 38±3 41±4 22±3 23±4 96±12 93±8 205±17 186±19 18±2 18±4 溶剂对照组 38±3 37±8 22±3 23±4 98±7 100±12 202±15 197±10 18±2 19±4 阳性对照组 881±18 876±35 900±11 1 077±111 964±113 996±8 1 024±37 1 011±8 392±8 392±22 2.2 染色体畸变试验结果
染色体分析结果显示,阳性对照组能够诱发受试细胞染色体的畸变率明显增高,24 h在+ S9和-S9的情况下染色体畸变率分别为11%和11%,与溶剂对照组相比,均有统计学差异(P<0.05);23.74、47.48和94.96 μg/ml受试物在24 h、+S9条件下染色体畸变率分别为1%、1%和0.5%,24 h、-S9条件下染色体畸变率分别为0%、0.5%和0%;4 h、-S9条件下染色体畸变率分别为0%、0%和0%。综上,受试物各剂量组细胞染色体畸变率均小于5%,与溶剂对照组结果相比,其差异均无统计学意义(P>0.05)。结果见表4~6。
表 4 WA对24 h体外培养CHO细胞的染色体畸变试验结果(+S9)组别 观察细胞数
(个)各类染色体畸变数 畸变细胞数
(个)畸变率
(%)断裂 断片 双着丝粒 三辐体 四辐体 碎片或微小体 环状 多倍体 23.74 μg/ml 200 1 1 0 0 0 0 0 1 2 1 47.48 μg/ml 200 2 2 0 0 0 0 0 0 2 1 94.96 μg/ml 200 0 0 0 0 0 0 0 0 1 0.5 溶剂对照组 200 200 0 0 0 0 0 3 3 3 1.5 阳性对照组 100 9 0 0 1 3 3 0 0 14 14 注:阳性对照组:+S9、环磷酰胺(60 mg/ml);溶剂对照组:DMSO;* P<0.05,与溶剂对照组比较。 表 5 WA对24 h体外培养CHO细胞的染色体畸变试验结果(-S9)组别 观察细胞数
(个)各类染色体畸变数 畸变细胞数
(个)畸变率
(%)断裂 断片 双着丝粒 三辐体 四辐体 碎片或微小体 环状 多倍体 23.74 μg/ml 200 0 0 0 0 0 0 0 0 0 0 47.48 μg/ml 200 1 0 0 0 0 0 0 0 1 0.5 94.96 μg/ml 200 0 0 0 0 0 0 0 0 0 0 溶剂对照组 200 0 0 0 0 0 0 0 0 0 0 阳性对照组 100 4 4 0 0 0 0 4 0 12 12* 注:阳性对照组:-S9、丝裂霉素C(0.5 mg/ml);溶剂对照组:DMSO;*P<0.05,与溶剂对照组比较。 表 6 WA对4 h体外培养CHO细胞的染色体畸变试验结果(-S9/4 h)组别 观察细胞数
(个)各类染色体畸变数 畸变细胞数
(个)畸变率
(%)断裂 断片 双着丝粒 三辐体 四辐体 碎片或微小体 环状 多倍体 23.74 μg/ml 200 0 0 0 0 0 0 0 0 0 0 47.48 μg/ml 200 0 0 0 0 0 0 0 0 0 0 94.96 μg/ml 200 0 0 0 0 0 0 0 0 0 0 溶剂对照组 200 阳性对照组 * P<0.05,与溶剂对照组比较。 2.3 小鼠骨髓微核试验的结果
试验结果经统计学分析表明,WA在100、200、400 mg/kg剂量下未观察到对小鼠骨髓的抑制作用,溶媒对照组和阳性对照组雌、雄性小鼠骨髓PCE微核发生率分别为2.10‰和21.36‰、1.90‰和20.88‰,两组相比差异均有统计学意义(P<0.05),验证了本次试验系统的有效性。WA 100和2 000 mg/kg剂量24 h采样组雌、雄小鼠骨髓PCE微核率分别为1.60‰和1.80‰、1.90‰和1.60‰;400 mg/kg剂量24 h和48 h采样组雌、雄小鼠骨髓PCE微核率分别为2.10‰和2.80‰、2.40‰和1.29‰,与溶媒对照组相比均无显著差异(P>0.05)。结果见表7。
表 7 WA对小鼠骨髓嗜多染红细胞的微核效应试验结果组别 性别 动物数(只) 观察PCE数(个) PCE/NCE(${\rm{\bar x}}$±s) 微核率(${\rm{\bar x}}$±s, ‰) 100 mg/kg 雌 5 10 020 2.40±0.89 1.60±0.96 200 mg/kg 雌 5 10 010 1.63±0.40 1.80±1.31 400 mg/kg (24 h采样) 雌 5 10 016 1.95±0.40 2.10±1.20 400 mg/kg (48 h采样) 雌 5 10 009 1.58±0.44 2.80±1.81 溶媒对照组 雌 5 10 009 2.37±0.69 2.10±1.20 阳性对照组 雌 5 9 095 2.18±0.30 21.36±7.84* 100 mg/kg 雄 5 10 016 1.56±0.30 1.90±1.28 200 mg/kg 雄 5 10 009 1.52±0.28 1.60±0.70 400 mg/kg (24 h采样) 雄 5 10 006 1.40±0.73 2.40±2.01 400 mg/kg (48 h采样) 雄 5 8 802 1.43±0.63 1.29±1.26 溶媒对照组 雄 5 9 029 2.13±0.58 1.90±1.45 阳性对照组 雄 5 10 011 1.62±0.42 20.88±4.94* *P<0.05,与溶剂对照组比较。 3. 讨论
沙门菌回复突变试验(Ames试验)被研究者广为采用,该法的特点是快速、简便、敏感、经济,是经典的测试化学物质或药物致突变性实验[10]。染色体畸变分析是采用中国仓鼠卵巢(CHO)细胞体外培养的方法进行的。CHO细胞在加或不加代谢活化系统的条件下,与受试物接触一定时间后再于收集染色体4 h前用秋水仙碱处理,使细胞的有丝分裂停止在中期相。然后收集细胞,经低渗、固定、涂片和染色后,在显微镜下观察染色体数量和结构的改变,检测受试物的诱变性[7]。微核试验是检测化合物对染色体损伤作用的重要方法。凡能使染色体发生断裂或使染色体和纺锤体联结损伤的化合物,微核试验都可检测[8]。
本研究采用遗传毒性研究经典组合的方法,分别从原核系统到真核系统,从体外试验系统到体内试验系统,体外试验中包含了加与不加代谢活化系统,能检测基因突变、染色体畸变等多个遗传学终点,符合国际标准化的要求[5-6]。
本研究结果显示,本试验条件下,采用标准平板掺入法,WA在每皿5 000、500、50、5、0.5 μg的受试剂量下,加或不加S9时对组氨酸缺陷型鼠伤寒沙门菌均无致突变性;对CHO细胞,在23.74、47.48、94.96 μg/ml 3个剂量组,加或不加S9,于24 h和48 h诱发的细胞染色体畸变率均小于5%,与溶剂对照组结果相比较其差异均无统计学意义(P>0.05),表明WA在受试剂量下无致CHO细胞染色体畸变效应;WA在100、200、400 mg/kg 3个剂量下,对ICR小鼠的微核诱发率与溶剂对照组比较均无显著差异,表明其在受试剂量下对ICR小鼠无致微核效应[14]。上述结果提示WA没有遗传毒性和潜在致癌性。
此前有文献表明WA对小细胞肺癌的增殖有抑制作用,但对于遗传毒性未见报道。本研究对降低WA在研发过程中用于临床试验和疾病治疗的用药风险发挥重要作用。
-
[1] BANERJEE K, RESAT H. Constitutive activation of STAT3 in breast cancer cells: a review[J]. Int J Cancer,2016,138(11):2570-2578. doi: 10.1002/ijc.29923 [2] QIN J J, YAN L, ZHANG J, et al. STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review[J]. J Exp Clin Cancer Res,2019,38(1):195. doi: 10.1186/s13046-019-1206-z [3] 孙思博, 金时代, 郭人花. STAT3在非小细胞肺癌耐药中的研究进展[J]. 中国肺癌杂志, 2019, 22(7):457-463. doi: 10.3779/j.issn.1009-3419.2019.07.08 [4] ZHANG W D, YU W Y, CAI G P, et al. A new synthetic derivative of cryptotanshinone KYZ3 as STAT3 inhibitor for triple-negative breast cancer therapy[J]. Cell Death Dis,2018,9(11):1098. doi: 10.1038/s41419-018-1139-z [5] 王雨辰, 陈越, 季鸣, 等. STAT3靶点抑制剂Bt354抗前列腺癌作用及其分子机制研究[J]. 药学学报, 2019, 54(10):1851-1857. [6] XU L Y, SHI L X, QIU S S, et al. Design, synthesis, and evaluation of cyanopyridines as anti-colorectal cancer agents via inhibiting STAT3 pathway[J]. Drug Des Devel Ther,2019,13:3369-3381. doi: 10.2147/DDDT.S217800 [7] DAI X X, YIN C T, ZHANG Y, et al. Osthole inhibits triple negative breast cancer cells by suppressing STAT3[J]. J Exp Clin Cancer Res,2018,37(1):322. doi: 10.1186/s13046-018-0992-z [8] MCHUGH J. Systemic sclerosis: STAT3 - A key integrator of profibrotic signalling[J]. Nat Rev Rheumatol,2017,13(12):693. doi: 10.1038/nrrheum.2017.190 [9] LIU L, LIU F B, HUANG M, et al. Circular RNA ciRS-7 promotes the proliferation and metastasis of pancreatic cancer by regulating miR-7-mediated EGFR/STAT3 signaling pathway[J]. HBPD INT,2019,18(6):580-586. [10] YU H, LEE H, HERRMANN A, et al. Revisiting STAT3 signalling in cancer: new and unexpected biological functions[J]. Nat Rev Cancer,2014,14(11):736-746. doi: 10.1038/nrc3818 [11] CHEN Q, LV J, YANG W W, et al. Targeted inhibition of STAT3 as a potential treatment strategy for atherosclerosis[J]. Theranostics,2019,9(22):6424-6442. doi: 10.7150/thno.35528 [12] TURKSON J, RYAN D, KIM J S, et al. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation[J]. J Biol Chem,2001,276(48):45443-45455. doi: 10.1074/jbc.M107527200 [13] TIMOFEEVA O A, TARASOVA N I, ZHANG X P, et al. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain[J]. Proc Natl Acad Sci USA,2013,110(4):1267-1272. doi: 10.1073/pnas.1211805110 [14] HUANG M, SONG K, LIU X Y, et al. AlloFinder: a strategy for allosteric modulator discovery and allosterome analyses[J]. Nucleic Acids Res,2018,46(W1):W451-W458. doi: 10.1093/nar/gky374 [15] HUANG W, DONG Z Z, WANG F, et al. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion[J]. ACS Chem Biol,2014,9(5):1188-1196. doi: 10.1021/cb500071v [16] BUETTNER R, CORZANO R, RASHID R, et al. Alkylation of cysteine 468 in Stat3 defines a novel site for therapeutic development[J]. ACS Chem Biol,2011,6(5):432-443. doi: 10.1021/cb100253e [17] CHENG X, LIU Y Q, WANG G Z, et al. Proteomic identification of the oncoprotein STAT3 as a target of a novel Skp1 inhibitor[J]. Oncotarget,2017,8(2):2681-2693. doi: 10.18632/oncotarget.13153 [18] SCHUST J, SPERL B, HOLLIS A, et al. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization[J]. Chem Biol,2006,13(11):1235-1242. doi: 10.1016/j.chembiol.2006.09.018 [19] CHEN H J, YANG Z D, DING C Y, et al. Discovery of O-alkylamino tethered niclosamide derivatives as potent and orally bioavailable anticancer agents[J]. ACS Med Chem Lett,2013,4(2):180-185. doi: 10.1021/ml3003082 [20] SHIN D S, KIM H N, SHIN K D, et al. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells[J]. Cancer Res,2009,69(1):193-202. doi: 10.1158/0008-5472.CAN-08-2575 [21] IWAMOTO K, UEHARA Y, INOUE Y, et al. Inhibition of STAT3 by anticancer drug bendamustine[J]. PLoS One,2017,12(1):e0170709. doi: 10.1371/journal.pone.0170709 [22] CHANG Y F, LIM K H, CHIANG Y W, et al. STAT3 induces G9a to exacerbate HER3 expression for the survival of epidermal growth factor receptor-tyrosine kinase inhibitors in lung cancers[J]. BMC Cancer,2019,19(1):959. doi: 10.1186/s12885-019-6217-9 [23] SHASTRI A, CHOUDHARY G, TEIXEIRA M, et al. Antisense STAT3 inhibitor decreases viability of myelodysplastic and leukemic stem cells[J]. J Clin Invest,2018,128(12):5479-5488. doi: 10.1172/JCI120156 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 10559
- HTML全文浏览量: 5304
- PDF下载量: 140
- 被引次数: 0
