

 下载:
下载:
 下载:
下载: 

</td></tr><tr><td class="table_top_border2" style="width:12%" align="center" valign="middle">一级指标</td><td class="table_top_border2" style="width:12%" align="center" valign="middle">二级指标</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="top">用药指征</td><td class="table_top_border2" align="center" valign="top">适应证</td><td class="table_top_border2" align="left" valign="top">适用于:①预防初次和重复使用致吐性肿瘤化疗(包括高剂量顺铂)引起的恶心和呕吐;②预防或治疗手术后恶心和/或呕吐。</td><td class="table_top_border2" align="center" valign="top">100</td></tr><tr><td align="center" valign="top"></td><td align="center" valign="top">禁忌证</td><td align="left" valign="top">已知对本品过敏的患者禁用。</td><td align="center" valign="top">100</td></tr><tr><td align="center" valign="top">用药过程</td><td align="center" valign="top">用法用量</td><td align="left" valign="top">①预防和治疗化疗引起的恶心呕吐:1.8 mg/kg 或是固定剂量100 mg;②预防或治疗手术后恶心和/或呕吐:12.5 mg。</td><td align="center" valign="top"> 80</td></tr><tr><td align="center" valign="top"></td><td align="center" valign="top">给药时间</td><td align="left" valign="top">①预防和治疗化疗引起的恶心呕吐:化疗前30 min给药;②预防手术后恶心和/或呕吐;麻醉停止前15 min;③治疗手术后恶心和/或呕吐:刚出现恶心、呕吐时。</td><td align="center" valign="top">100</td></tr><tr><td align="center" valign="top"></td><td align="center" valign="top">溶媒</td><td align="left" valign="top">预防和治疗化疗引起的恶心呕吐:溶于50 ml 溶媒(0.9%氯化钠注射液或是5%葡萄糖注射液)静脉输注15 min 以上。</td><td align="center" valign="top"> 80</td></tr><tr><td align="center" valign="top"></td><td align="center" valign="top">特殊人群</td><td align="left" valign="top">①儿童:尚无2岁以下患儿使用本品经验,2~17岁患儿耐受性好;<br>②妊娠期:尚缺乏在怀孕妇女中的充分的良好对照的研究;<br>③哺乳期:本品是否乳汁排泄尚不清楚,慎用;<br>④65岁以上患者不需要调整剂量,但用药仍须谨慎;<br>⑤肾功能不全:无需调整剂量;<br>⑥肝功能不全:无需调整剂量。</td><td align="center" valign="top">100</td></tr><tr><td align="center" valign="top"></td><td align="center" valign="top">疗程</td><td align="left" valign="top">化疗期间或是化疗后,存在恶心呕吐感即可。</td><td align="center" valign="top">100</td></tr><tr><td align="center" valign="top"></td><td align="center" valign="top">联合用药</td><td align="left" valign="top">单药不能控制的恶心呕吐,可联合使用激素地塞米松,严重的可再联合NK-1抑制剂阿瑞吡坦。</td><td align="center" valign="top">100</td></tr><tr><td align="center" valign="top"></td><td align="center" valign="top">相互作用</td><td align="left" valign="top">本品不能与其它药物混合使用,输注后要冲洗输液管道。</td><td align="center" valign="top">100</td></tr><tr><td align="center" valign="top">用药结果</td><td align="center" valign="top">不良反应</td><td align="left" valign="top">发生本品不良反应停用本品并对症处理,上报不良反应相关信息至ADR监测系统和药剂科。</td><td align="center" valign="top">100</td></tr><tr><td align="center" valign="top"></td><td align="center" valign="top">疗效</td><td align="left" valign="top">恶心呕吐次数减少或消失。</td><td align="center" valign="top"> 90</td></tr><tr><td class="table_bottom_border" align="center" valign="top">管理指标</td><td class="table_bottom_border" align="center" valign="top">病程记录</td><td class="table_bottom_border" align="left" valign="top">①病程录中有相关的用药指证;<br>②联合用药以及对患者恶心呕吐严重程度的描述。</td><td class="table_bottom_border" align="center" valign="top"> 90</td></tr></tbody>
</table></div></foreignObject></svg>)
-
多拉司琼是第一代5-HT3受体拮抗剂,可用于预防和治疗手术引起的恶心呕吐(PONV)和预防化学抗肿瘤治疗引起的恶心呕吐(CINV)。多拉司琼的代谢产物具有可阻滞钠通道和5-HT3受体的活性,在手术和化疗过程中常作为预防和治疗恶心呕吐的主要药物,指南中推荐根据抗肿瘤药物的致吐级别调整止吐用药的剂量和疗程[1]。但是在临床使用过程中存在着不合理使用情况,我们参照美国医院药师协会(ASHP)制定的药物利用评价(DUE)方法[2-4],建立多拉司琼药物利用评价标准,对我院多拉司琼的使用情况进行回顾性分析,以期为规范多拉司琼的临床用药提供参考。
-
我院2021年1月至2021年6月使用多拉司琼的出院患者共2 382例,根据院内处方审核软件SQL SERVER 2008数据库自带随机生成数字序列随机抽取900例,剔除患者信息及病历记录不全者,共收集有效病例794例。其中男性374例(占比47.1%);女性420例(占比52.9%);特殊人群235例,包括老年人182例,肾功能不全患者17例,肝功能不全36例。
-
参照ASHP对DUE标准操作指南为标准基础,根据多拉司琼药品说明书、《NCCN止吐指南》(2020.V1版)、《新编药物学》(2017版)、《临床用药须知》(2010版)及其它相关文献资料[5-6]草拟多拉司琼DUE标准及预期目标值,经我院药事管理和药物治疗学委员会合理用药组成员(由高级职称的临床医生和药学专家组成)逐条从科学性、实用性、可行性方面进行讨论,并结合医院实际情况修订后,确立多拉司琼DUE标准。
多拉司琼DUE标准中的评价项目包括用药指征、用药过程、用药结果和管理指标等4个一级指标,下设适应证、禁忌证、用法用量、给药时间、溶媒、特殊人群、疗程、联合用药、相互作用、不良反应、疗效、病程记录等12个二级指标。本文中用于评价多拉司琼DUE评价标准的各因素均为相同重要,因此未予以权重比例的区分。多拉司琼DUE标准见表1。
表 1 多拉司琼DUE标准
评价指标 标准内容 预期目标值(%) 一级指标 二级指标 用药指征 适应证 适用于:①预防初次和重复使用致吐性肿瘤化疗(包括高剂量顺铂)引起的恶心和呕吐;②预防或治疗手术后恶心和/或呕吐。 100 禁忌证 已知对本品过敏的患者禁用。 100 用药过程 用法用量 ①预防和治疗化疗引起的恶心呕吐:1.8 mg/kg 或是固定剂量100 mg;②预防或治疗手术后恶心和/或呕吐:12.5 mg。 80 给药时间 ①预防和治疗化疗引起的恶心呕吐:化疗前30 min给药;②预防手术后恶心和/或呕吐;麻醉停止前15 min;③治疗手术后恶心和/或呕吐:刚出现恶心、呕吐时。 100 溶媒 预防和治疗化疗引起的恶心呕吐:溶于50 ml 溶媒(0.9%氯化钠注射液或是5%葡萄糖注射液)静脉输注15 min 以上。 80 特殊人群 ①儿童:尚无2岁以下患儿使用本品经验,2~17岁患儿耐受性好;
②妊娠期:尚缺乏在怀孕妇女中的充分的良好对照的研究;
③哺乳期:本品是否乳汁排泄尚不清楚,慎用;
④65岁以上患者不需要调整剂量,但用药仍须谨慎;
⑤肾功能不全:无需调整剂量;
⑥肝功能不全:无需调整剂量。100 疗程 化疗期间或是化疗后,存在恶心呕吐感即可。 100 联合用药 单药不能控制的恶心呕吐,可联合使用激素地塞米松,严重的可再联合NK-1抑制剂阿瑞吡坦。 100 相互作用 本品不能与其它药物混合使用,输注后要冲洗输液管道。 100 用药结果 不良反应 发生本品不良反应停用本品并对症处理,上报不良反应相关信息至ADR监测系统和药剂科。 100 疗效 恶心呕吐次数减少或消失。 90 管理指标 病程记录 ①病程录中有相关的用药指证;
②联合用药以及对患者恶心呕吐严重程度的描述。90 -
多拉司琼DUE评价结果(表2)显示,随机抽取的794例病例中,105例完全符合标准,占比13.23%。主要存在的不合理用药情况有:适应证不符24例(占比3.0%);用于治疗和预防PONV用法用量不符合标准要求258例,治疗和预防CINV用法用量不符合标准要求186例。794例全部选择使用0.9%氯化钠注射液,其中188例(23.7%)溶媒体积选择50 ml,符合标准。在调查期间,未见关于多拉司琼的不良反应报告,病历中也未有不良反应记录;无相互作用;治疗有效符合标准率100%;病程记录符合标准率68.0%。
表 2 多拉司琼DUE评价结果
评价指标 标准符合率% 一级指标 二级指标 用药指征 适应证 96.9 禁忌证 100.0 用药过程 用法用量 43.3 给药时间 100.0 溶媒 23.7 特殊人群 100.0 疗程 100.0 联合用药 86.6 相互作用 100.0 用药结果 不良反应 100.0 疗效 100.0 管理指标 病程记录 68.0 -
多拉司琼的适应证明确,794份病例中符合用药指征标准的有770例(占比为96.9%),不符合标准的24例中,有10例是手术日前两日开始使用多拉司琼1剂,9例患者是既未进行手术治疗也未进行化疗便使用多拉司琼1剂,5例是手术后4d~5d使用多拉司琼1剂,且上述24例患者病程中均未记录恶性、呕吐症状。在抽取的病例中不存在禁忌证用药。
-
在抽取的病例中,有113例用药过程完全符合标准,占比仅14.2%;在用法用量方面,258例用于术后治疗恶心呕吐的病例存在超剂量使用情况,标准剂量应为12.5 mg,56例使用25 mg,130例使用50 mg,72例使用100 mg;在预防和治疗CINV中,186例存在用药剂量不足的情况,标准剂量应为100 mg或是1.8 mg/kg,8例使用87.5 mg,17例使用62.5 mg,这25例患者体重均超过50 kg,161例使用50 mg;溶媒选择的标准符合率只有23.7%,其中33例选择250 ml 0.9%氯化钠注射液,566例选择100 ml 0.9%氯化钠注射液,均不符合要求;只有195例是选择加入50 ml 0.9%氯化钠注射液静脉输注或是直接静脉推注给药,符合要求;用药时间、特殊人群和相互作用的符合标准率是100%,联合用药符合标准率是86.6%,不符合联合用药标准的有105例,在化疗中预防和治疗CINV时,不仅使用多拉司琼,还联合使用8 mg昂丹司琼注射液。
-
本研究中,使用多拉司琼症状好转的有效率为100%,在这些病例的病史记录中,术后或是化疗后,在使用多拉司琼后未出现明显的恶心呕吐症状,并且也没有升级使用第二代5-HT3抑制剂或是神经激肽1(NK-1)抑制剂。
在抽取的病例中,病程记录中未记录发生多拉司琼相关药品不良反应,也不排除有发生的不良反应未记录在病程记录中。在抽取病例期间,药剂科没有接收到该药品相关不良反应事件的上报信息。
-
在抽取的病例中,不符合标准的病程记录254份,主要问题是没有写明是用于预防或是治疗PONV或是CINV。
-
我院正在不断地加强药品合理使用管理,加强对辅助用药合理使用的评价。在对抽取病例进行评价中发现,我院在使用多拉司琼的临床治疗过程中存在多处不合理现象,包括用药指征中的无适应证用药、用药剂量不符等。在此次抽取病例中存在超剂量使用的情况中,尚未出现不良反应,但根据文献报道,超剂量使用多拉司琼后患者可出现QT间期延长的不良反应[7]。在不足剂量使用的情况下,会影响药物的疗效,降低患者的生活质量,需要延长给药时间或是换用二代5-HT3抑制剂或是联合其他药理作用的止吐药物,这将会增加患者的经济负担和医保支出[8]。
研究中发现的严重不合理用药问题,是联合使用两种5-HT3拮抗剂,如多拉司琼联合昂丹司琼,两者同属第一代5-HT3拮抗剂,属于重复用药。在临床实际用药中,尤其是对于抗肿瘤治疗,存在单一的5-HT3拮抗剂不能有效控制的CINV,临床上根据化疗方案的致吐级别和患者的耐受能力,联合使用其他药理作用机制的药物,如NK-1抑制剂阿瑞匹坦、地塞米松,PPI制剂或是多巴胺受体拮抗剂甲氧氯普胺等。
其次的不合理问题是溶媒体积的选择。根据多拉司琼的药物动力学参数[9],在手术麻醉结束前15 min或是化疗前30 min给药,可以较好的发挥止吐作用。一般建议静脉推注或是加入50 ml 0.9%氯化钠注射液静脉输注,输注时间为10 min左右。目前临床医生大多数选择100 ml的溶媒,主要是因为100 ml溶媒常规输注时间是30 min,在多拉司琼注射结束后就可以更换至化疗药物输注,正好符合预防止吐药物在化疗前30 min使用的时间窗口。如果输液体积是250 ml,则延长了5倍的输注时间,这就改变多拉司琼在体内的药动学特征,影响药物的疗效[10]。
通过使用DUE合理用药标准,抽样分析点评多拉司琼的用药情况,掌握具体科室和医生的不合理用药情况,做到及时发现问题,分析问题,将问题反馈至临床和行政管理部门,可以针对性地进行沟通交流,更好地协调处理用药不合理问题,减少不合理用药的发生。将DUE作为一种用药评价模式,成为临床药师评价临床药物治疗的工具,分析并解决问题,提高临床用药合格率,以保证药物使用合理、安全和有效,减少患者不必要的经济负担和医保支出。目前住院系统药物医嘱审核存在滞后问题,是采用回顾性评价住院药物医嘱,根据药品使用数据异常筛查并针对性点评药品,根据药品点评结果和行政部门与临床使用科室进行沟通,后续对相同药品进行再次点评,复核整改效果。针对审核滞后问题。我院正在考虑上线实时住院医嘱审核,可将以上DUE标准转化至智能审核系统,将不合理用药情况在医生开具医嘱时进行提醒和拦截,提高药品的规范合理使用。但因受限于技术和人力原因,暂时未能上线。
临床药师作为临床药物治疗合理用药工作的成员之一,在使用该评价标准对多拉司琼进行评价分析时,存在疗效不能通过辅助检查的数字化指标量化对比分析的问题,只能将患者的疗效反馈通过病程记录的内容反馈在病历中,在以后的指标设计中,应采取更为可靠、客观的综合评价方法对指标进行验证,设计更科学合理的评价方法,可以更好的推广药物合理使用评价标准的方法。
Establishment and application of DUE criteria of Dolasetron
-
摘要:
目的 通过药物利用评价(DUE)方法评价复旦大学附属华东医院多拉司琼临床应用情况,为促进临床合理使用多拉司琼提供参考。 方法 随机抽取医院2021年1月至2021年6月使用多拉司琼的病例794例,依据建立的DUE标准进行回顾性分析。 结果 建立了多拉司琼DUE标准,包括用药指征、用药过程、用药结果和管理指标4个部分。 结论 建立的多拉司琼DUE标准具有较好的实用性,在临床实践应用中可发现临床用药过程中存在的问题或不足,对促进临床合理用药具有重要指导意义。 Abstract:Objective To establish the drug use evaluation ( DUE) of Dolasetron, evaluate the rationality of the clinical use of Dolasetron and provide a reference for the rationally clinical use of Dolasetron. Methods On the basis of Dolasetron DUE criteria, a retrospective analysis was made in 794 hospitalized patients from January 2021 to June 2021. Results The drug use evaluation criterion on Dolasetron consisted of drug indications, drug use process, the result of drug use and indication management. Conclusion There are some inappropriate medication problems in Dolasetron utilization in the hospital. The DUE criterion is very practical which could be used to standardize the clinical utilization of Dolasetron. -
Key words:
- Dolasetron /
- drug use evolution /
- drug use evaluation criteria /
- rational use of drug
-
放线菌以其能产生结构新颖且有良好生物活性的先导化合物而备受关注[1],一直被认为是天然药物的重要生产者,其主要结构类型包括聚酮、生物碱、多肽和萜烯类化合物等,同时涵盖了多种多样的生物活性如抗菌、抗寄生虫、免疫调节、抗炎、抗癌等[2-4],这突显了放线菌具有不可预估的药物开发潜力。
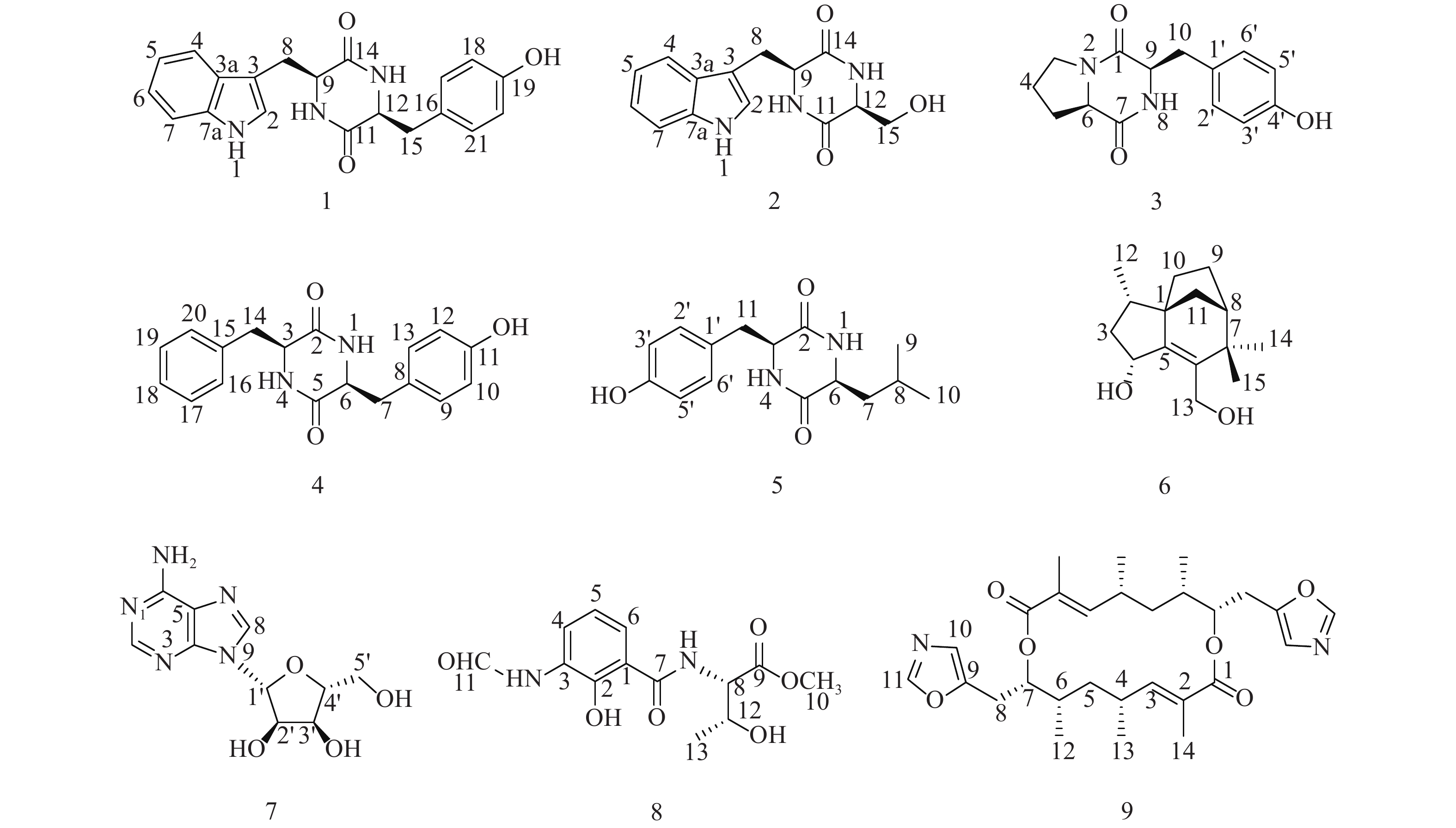
随着研究的深入,陆地和普通环境中的资源日趋枯竭,很多微生物及其次生代谢产物被重复开发和提取分离,发现新活性分子的几率愈来愈低,开发创新药物的难度越来越大[5-6];而极地极端的生态环境造就的微生物具有产生更为特别的化学骨架和活性次生代谢产物的能力,是新型药源分子的重要来源。本文以采自北极楚克奇海域海绵共附生放线菌Streptomyces sp. LHW11-07为研究对象,从其发酵浸膏中分离鉴定了9个单体化合物1~9(图1),其中化合物1和2为该属内首次分离得到。
1. 材料和方法
1.1 实验仪器与试剂
AMX-600型核磁共振仪(德国Bruker公司);Xevo G2-XS Q-TOF液质联用仪、1525/2996, 2998型高效液相色谱仪(美国Waters公司);半制备型HPLC色谱柱(Atlantis Prep T3,美国Waters公司;YMC C18,日本YMC公司);中压柱色谱仪(法国Interchim公司);恒温振荡培养箱(上海一恒科学仪器有限公司);N-1000型旋转蒸发仪(上海爱郎仪器有限公司);反相ODS硅胶和Sephadex LH-20柱色谱填料(Pharmacia公司);正相硅胶(200-300目)和TLC薄层板(烟台江友硅胶开发有限公司);分析级试剂(上海化学试剂公司);色谱级试剂(德国Merck公司);氘代试剂(美国剑桥同位素实验室公司)。
1.2 菌株的来源及鉴定
菌株分离于北极海域来源的海绵样本,经16S rRNA基因序列鉴定为Streptomyces sp.,编号为LHW11-07,菌种保存于上海交通大学医学院附属仁济医院药学部海洋药物研究中心。
1.3 菌株的大发酵
培养基为ISP2:葡萄糖(4 g/L)、酵母提取物(4 g/L)、麦芽糖提取物(10 g/L)以及海盐(25 g/L),加水溶解后调节pH为7.2~7.4,分装后高压灭菌20 min (121 ℃),冷却备用。
挑取Streptomyces sp. LHW11-07单菌落至1级种子培养基里(100 ml ISP2培养基至250 ml三角瓶),置于30 ℃,220 r/min的恒温摇床培养3 d,得1级种子液;将1级种子液按5%接种量接到2级种子培养基里(150 ml ISP2培养基至500 ml三角瓶),置于30 ℃,220 r/min的恒温摇床培养3 d,得2级种子液;将2级种子液按5%接种量接到大发酵培养基里(700 ml ISP2培养基至2 L三角瓶),置于30 ℃,220 r/min的恒温摇床培养7 d,共得到发酵液50 L。
1.4 发酵产物的提取与分离
菌株培养7 d后,用等体积的乙酸乙酯萃取3次,合并乙酸乙酯提取液,减压浓缩得粗浸膏9.8 g。粗浸膏先经凝胶柱分离,二氯甲烷:甲醇(1∶1)的混合溶剂进行洗脱,得到组份Fr.1~Fr.7。
组份Fr.4经正相中压柱色谱分离(二氯甲烷:甲醇100:0~0:100),得到组份Fr.4a~Fr.4j。组份Fr.4d再经反相中压柱色谱分离(10%~100%乙腈水),得到组份Fr.4d1~Fr.4d8,Fr.4d2和Fr.4d3用反相半制备HPLC纯化(35%甲醇水,YMC C18),分别得到化合物1 (12 mg, tR=26 min)和2 (47.6 mg, tR=30 min);Fr.4d6用反相半制备HPLC纯化(49%甲醇水,YMC C18),得到化合物3 (8.4 mg, tR=23 min)和4 (2.0 mg, tR=30 min);组份Fr.4g再经凝胶柱纯化,洗脱剂为正己烷:二氯甲烷:甲醇(4∶5∶1),得到组份Fr.4g1~Fr.4g11,其中Fr.4g3用反相半制备HPLC纯化(25%乙腈水,YMC C18),得到化合物5 (1.2 mg, tR=18 min)。
组份Fr.7经反相中压柱色谱分离(10%~100%乙腈水),得到组份Fr.7A~Fr.7D。组份Fr.7A用反相半制备HPLC纯化(20%乙腈水,YMC C18),得到化合物6 (18 mg, tR=19 min)和7 (3.4 mg, tR=25 min);组份Fr.7B经正相中压柱分离(石油醚:丙酮100:0~0:100),得到组份Fr.7B1~Fr.7B9,Fr.7B4用反相半制备HPLC纯化(88%乙腈水,Atlantis Prep T3),得到化合物8 (12 mg, tR=23 min)和9 (8.4 mg, tR=26 min)。
2. 结构鉴定
化合物1:淡黄色固体,ESI-MS显示准分子离子峰m/z 372 [M+Na]+。1H-NMR (600 MHz, DMSO)显示在低场区有吲哚环的特征信号δH 7.00 (1H, s, H-2),7.49 (1H, d, J=8.0 Hz, H-4),7.02 (1H, t, J=7.6 Hz, H-5),7.05 (1H, t, J=7.6 Hz, H-6),7.32 (1H, d, J=8.0 Hz, H-7);4个活泼氢质子信号δH 10.89 (1H, d, J=2.6 Hz, NH-1),7.83 (1H, d, J=3.0 Hz, NH-10),7.62 (1H, d, J=3.0 Hz, NH-13),9.20 (1H, s, OH-19);4个芳香质子信号δH 6.53 (2H, d, J=8.4 Hz, H-17, H-21),6.59 (2H, d, J=8.4 Hz, H-18, H-20),提示分子中有1个对位二取代的苯环;在高场区有两组亚甲基质子信号δH 2.80 (1H, dd, J=14.5, 4.5 Hz, H-8),2.43 (1H, ov, H-8),δH 1.83 (1H, dd, J=13.4, 6.9 Hz, H-15),2.47 (1H, ov, H-15),两个次甲基质子信号δH 4.01 (1H, m, H-9),3.95 (1H, m, H-12)。13C-NMR (150 MHz, DMSO)结合DEPT谱表明其有20个碳信号,2个酮羰基碳δC 166.7,166.2,14个芳香碳,2个亚甲基碳δC 30.0,40.0和2个次甲基碳δC 55.9,55.2。对其碳信号进行归属:δC 118.7 (C-2)、108.9 (C-3)、127.5 (C-3a)、118.4 (C-4)、120.8 (C-5)、124.3 (C-6)、111.3 (C-7)、136.0 (C-7a)、30.0 (C-8)、55.9 (C-9)、166.7 (C-11)、55.2 (C-12)、166.2 (C-14)、40.1 (C-15)、126.4 (C-16)、130.7 (C-17, C-21)、114.9 (C-18, C-20)、156.0 (C-19)。以上数据与文献[7]对比基本一致,故确定为cyclo-(L-Tyr-L-Trp)。
化合物2:淡黄色固体,ESI-MS显示准分子离子峰m/z 274 [M+H]+。1H-NMR (600 MHz, DMSO)发现其与化合物1一样有吲哚环的特征信号δH 7.09 (1H, s, H-2),7.52 (1H, d, J = 7.9 Hz, H-4),6.99 (1H, t, J = 7.8 Hz, H-5),7.02 (1H, t, J = 7.8 Hz, H-6),7.30 (1H, d, J = 7.8 Hz, H-7);3个活泼氨基质子信号δH 10.87 (1H, s, NH-1),δH 8.30 (1H, m, NH-10),7.85 (1H, d, J = 2.9 Hz, NH-13);两组亚甲基质子信号δH 3.21 (1H, m, H-8),3.13 (1H, m, H-8),δH 3.65 (1H, m, H-15),3.05 (1H, m, H-15),两个次甲基质子信号δH 4.87 (1H, m, H-9),4.00 (1H, m, H-12)。13C-NMR (150 MHz, DMSO)结合DEPT谱表明其有14个碳信号,2个酮羰基碳δC 167.2,165.7,8个芳香碳,2个亚甲基碳δC 63.0,30.3和2个次甲基碳δC 57.3,55.5。对其碳信号进行归属:δC 127.6 (C-2)、111.2 (C-3)、136.0 (C-3a)、118.6 (C-4)、120.8 (C-5)、124.0 (C-6)、118.3 (C-7)、109.0 (C-7a)、30.3 (C-8)、57.3 (C-9)、167.2 (C-11)、55.5 (C-12)、165.7 (C-14)、63.0 (C-15)。以上数据与文献[8]对比基本一致,故确定为cyclo-(L-Trp-L-Ser)。
化合物3:白色固体,ESI-MS显示准分子离子峰m/z 261 [M+H]+。1H-NMR (600 MHz, DMSO)提示有2个活泼氢质子信号δH 7.87 (1H, s, NH-8),9.22 (1H, s, OH-4’);1组对位二取代的苯环芳香质子信号δH 7.04 (2H, d, J = 8.2 Hz, H-2’, H-6’),6.63 (2H, d, J = 8.2 Hz, H-3’, H-5’);2个次甲基质子信号δH 4.24 (1H, t, J = 8.2 Hz, H-6),4.03 (1H, dd, J = 9.9, 2.9 Hz, H-9);4组亚甲基质子信号δH 3.42 (1H, m, H-3),3.24 (1H, m, H-3),1.73 (2H, m, H2-4),2.00 (1H, m, H-5),1.41 (1H, m, H-5),2.93 (2H, m, H2-10)。13C-NMR (150 MHz, DMSO)结合DEPT谱表明其有14个碳信号,2个酮羰基碳δC 168.9,165.1,6个芳香碳,2个次甲基碳δC 58.4,56.0以及4个亚甲基碳δC 44.6,34.7,27.8,21.9。对其碳信号进行归属:δC 165.1 (C-1)、44.6 (C-3)、21.9 (C-4)、27.8 (C-5)、58.4 (C-6)、168.9 (C-7)、56.0 (C-9)、34.7 (C-10)、127.0 (C-1’)、130.8 (C-2’, C-6’)、114.8 (C-3’, C-5’)、155.9 (C-4’)。以上数据与文献[9]对比基本一致,故确定为cyclo-(D-Tyr-D-Pro)。
化合物4:白色固体,ESI-MS显示准分子离子峰m/z 311 [M+H]+。1H-NMR (600 MHz, DMSO)显示3个活泼氢质子信号δH 9.30 (1H, s, OH-11),7.84 (2H, t, J = 2.9 Hz, NH-1, NH-4);9个芳香区质子信号:4个归为1组对位二取代苯环δH 6.85 (2H, d, J = 8.5 Hz, H-9, H-13),6.65 (2H, t, J = 8.5 Hz, H-10, H-12),5个归为1组单取代苯环δH 7.20 (1H, t, J = 7.6 Hz, H-18),7.04 (2H, d, J = 6.9 Hz, H-16, H-20),7.28 (2H, t, J = 7.6 Hz, H-17, H-19);2组亚甲基质子信号δH 2.58 (1H, dd, J = 13.6, 5.0 Hz, H-7),2.20 (1H, d, J = 6.5 Hz, H-7),2.19 (2H, dd, J = 13.6, 6.5 Hz, H2-14),2个次甲基质子信号δH 3.95 (1H, m, H-3),3.90 (1H, m, H-6)。13C-NMR (150 MHz, DMSO)结合DEPT谱显示其有18个碳信号,2个酮羰基碳δC 166.2,166.2,12个芳香碳,2个亚甲基碳δC 40.1,38.5和2个次甲基碳δC 55.7,55.4。对其碳信号进行归属:δC 166.2 (C-2)、55.7 (C-3)、166.3 (C-5)、55.4 (C-6)、40.1 (C-7)、126.5 (C-8)、130.8 (C-9, C-13)、115.0 (C-10, C-12)、156.1 (C-11)、38.5 (C-14)、136.7 (C-15)、129.7 (C-16, C-20)、128.2 (C-17, C-19)、126.4 (C-18)。以上数据与文献[10]对比基本一致,故确定为cyclo-(L-Tyr-L-Phe)。
化合物5:白色固体,ESI-MS显示准分子离子峰m/z 277 [M+H]+。1H-NMR (600 MHz, DMSO)显示3个活泼氢质子信号δH 9.22 (1H, s, OH-4’),8.02 (2H, dd, J = 5.6, 2.5 Hz, NH-1, NH-4);4个芳香质子信号δH 6.90 (2H, d, J = 8.2 Hz, H-2’, H-6’),6.64 (2H, d, J = 8.2 Hz, H-3’, H-5’),提示分子中有1个对位二取代苯环;3个次甲基质子信号δH 4.06 (1H, q, J = 3.3 Hz, H-3),3.44 (1H, m, H-6),1.43 (1H, ov, H-8),2个亚甲基质子信号δH 1.43 (1H, m, H-7),1.23 (1H, m, H-7),2.69 (1H, q, J = 13.6, 4.8 Hz, H-11),3.01 (1H, q, J = 13.7, 3.7 Hz, H-11)以及2个末端甲基质子信号δH 0.63 (6H, ov, H3-9, H3-10)。13C-NMR (150 MHz, DMSO)结合DEPT谱显示其有15个碳信号,2个酮羰基碳δC 166.2,167.4,6个芳香碳,2个亚甲基碳δC 43.7,37.7,3个次甲基碳δC 55.7,52.3,21.4以及2个甲基碳δC 22.9,22.8。对其碳信号进行归属:δC 166.2 (C-2)、55.7 (C-3)、167.4 (C-5)、52.3 (C-6)、43.7 (C-7)、21.4 (C-8)、22.9 (C-9)、22.8 (C-10)、37.7 (C-11)、125.8 (C-1’)、131.2 (C-2’, C-6’)、114.8 (C-3’, C-5’)、156.4 (C-4’)。以上数据与文献[11]对比基本一致,故确定为cyclo-(L-Tyr-L-Leu)。
化合物6:白色固体,ESI-MS显示准分子离子峰m/z 259 [M+Na]+。1H-NMR (600 MHz, DMSO)显示有2个活泼氢质子信号δH 5.05 (1H, d, J = 4.5 Hz, OH-4)和4.39 (1H, q, J = 4.0 Hz, OH-13),3个甲基质子信号δH 0.88 (3H, d, J = 6.8 Hz, H3-12),0.98 (3H, s, H3-14)和δH 1.05 (3H, s, H3-15),5对亚甲基质子信号δH 2.14 (1H, m, H-3),1.23 (1H, m, H-3),1.74 (1H, m, H-9),1.60 (1H, m, H-9),1.44 (3H, m, H2-10, H-11),1.32 (1H, d, J = 10.4 Hz, H-11),3.95 (2H, m, H2-13),3个次甲基质子信号δH 1.68 (1H, m, H-2),4.57 (1H, m, H-4),1.77(1H, m, H-8)。13C-NMR (150 MHz, DMSO)结合DEPT谱共显示有15个碳信号,包括4个季碳δC 52.1,150.1,136.7,39.8;3个次甲基碳δC 35.2,69.8,46.4;5个亚甲基碳δC 42.4,23.8,28.7,36.4,57.1以及3个甲基碳δC 13.7,29.1,24.4。对其碳信号进行归属:δC 52.1 (C-1)、35.2 (C-2)、42.4 (C-3)、68.9 (C-4)、150.1 (C-5)、136.7 (C-6)、39.8 (C-7)、46.4 (C-8)、23.8 (C-9)、28.7 (C-10)、36.4 (C-11)、13.7 (C-12)、57.1 (C-13)、29.1 (C-14)、24.4 (C-15)。以上数据与文献[12]对比基本一致,故确定为albaflavenol B。
化合物7:白色结晶固体,ESI-MS显示准分子离子峰m/z 268 [M+H]+。1H-NMR (600 MHz, DMSO)可看出其有13个氢信号,包括5个活泼氢质子信号δH 3.56 (2H, m, NH2),5.49 (1H, s, OH-5’),5.36 (1H, t, J = 4.9 Hz, OH-2’)和5.23 (1H, s, OH-3’),4个连氧次甲基质子信号δH 4.56 (1H, s, H-2’),4.14 (1H, s, H-3’),3.96 (1H, m, H-4’)和5.90 (1H, d, J = 5.8 Hz, H-1’),1组亚甲基信号δH 3.66 (2H, m, H2-5’)以及2个低场区的烯氢质子信号δH 8.37 (1H, s, H-8)和8.21 (1H, s, H-2)。13C-NMR (150 MHz, DMSO)显示其共有10个碳信号,结合DEPT谱可推测有3个芳香季碳δC 149.9,119.8,154.3,2个连氮的芳香次甲基碳δC 151.7,138.6,4个次甲基碳δC 87.8,73.5,70.5,85.7,1个亚甲基碳δC 61.5。对其碳信号进行归属:δC 151.7 (C-2)、149.9 (C-4)、119.8 (C-5)、154.3 (C-6)、138.6 (C-8)、87.8 (C-1’)、73.5 (C-2’)、70.5 (C-3’)、85.7 (C-4’)、61.5 (C-5’)。以上数据与文献[13]对比基本吻合,故确定为β-adenosine。
化合物8:绿色无定型固体,ESI-MS显示准分子离子峰m/z 297 [M+H]+。1H-NMR (600 MHz, MeOD)显示有12个氢信号,包括1个活泼氢质子信号δH 8.37 (1H, s, H-11),1个甲氧基质子信号δH 3.79 (3H, s),1个甲基质子信号δH 1.25 (3H, d, J = 6.4 Hz, H3-13),3个低场区的芳香氢质子信号δH 8.31 (1H, d, J = 7.8 Hz, H-4),6.92 (1H, t, J = 8.0 Hz, H-5)和7.65 (1H, d, J = 8.0 Hz, H-6),以及2个次甲基质子信号δH 4.74 (1H, d, J = 3.2 Hz, H-8)和4.40 (1H, m, H-12),后与文献[14]对比发现其有4个活泼氢质子信号没有显示出来,而根据相关化学位移可确定其是同一个已知化合物。13C-NMR (150 MHz, MeOD)显示有13个碳信号,结合DEPT谱可推测有6个芳香碳δC 123.4,119.4,126.2,128.2,152.5,115.6,2个羰基碳δC 171.8,172.4,而δC 162.1为醛基碳,2个次甲基碳δC 59.4,68.4,1个甲基碳δC 20.5,以及1个甲氧基碳δC 52.9。对其碳信号进行归属:δC 115.6 (C-1)、152.5 (C-2)、128.2 (C-3)、126.2 (C-4)、119.4 (C-5)、123.4 (C-6)、171.8 (C-7)、59.4 (C-8)、172.4 (C-9)、52.9 (C-10)、162.1 (C-11)、68.4 (C-12)、20.5 (C-13)。以上数据与文献[14]对比基本吻合,故确定为N-formylantimyic acid methyl ester。
化合物9:白色粉末状固体,ESI-MS显示准分子离子峰m/z 499 [M+H]+。1H-NMR (600 MHz, DMSO)显示有19个氢信号:δH 8.20 (1H, s, H-11),6.82 (1H, s, H-10),6.32 (1H, dd, J = 10.5, 1.3 Hz, H-3),5.01 (1H, m, H-7),2.99 (1H, dd, J = 2.5, 15.8 Hz, H-8),2.80 (1H, dd, J = 10.3, 15.6 Hz, H-8),2.58 (1H, m, H-4),1.66 (1H, m, H-5),1.65 (3H, s, 2-Me),1.27 (1H, m, H-6),1.25 (1H, m, H-5),1.06 (3H, d, J = 6.5 Hz, 4-Me)和0.95 (3H, d, J = 5.9 Hz, 6-Me)。而13C-NMR (150 MHz, DMSO)结合DEPT谱显示只有14个碳信号:δC 166.0 (C-1),126.8 (C-2),147.4 (C-3),30.7 (C-4),37.6 (C-5),35.1 (C-6),74.5 (C-7),24.0 (C-8),149.4 (C-9),122.9 (C-10),151.3 (C-11),12.7 (2-Me),21.1 (4-Me)和16.2 (6-Me),说明这个化合物可能是一个具有对称结构的二聚体,通过与文献[15]中化合物conglobatin A对比后发现两者波谱数据完全吻合,故最终确定为conglobatin A。
3. 讨论
自上世纪发现青霉素以来,微生物中活性次生代谢产物一直是药物先导化合物的重要来源之一,据统计1940年—2019年间,科学家从微生物中开发出293种治疗不同疾病的临床药物[16]。但随着研究的深入,很多微生物及其次生代谢产物存在被重复开发和提取分离的问题,加之多重耐药性的产生,迫使人们需要开拓新的制造药物的微生物来源[5-6],而其中极地微生物资源是珍贵而特殊的。来自极地海洋等特殊生态环境的生物往往具有比陆地生物更为丰富的代谢途径和功能基因簇,增加了产生结构新颖且功能独特的次生代谢物的可能性。极地生物以微生物和一些能适应极端条件的海洋生物为主,然而与已报道的大量极地微生物相比,鲜有微生物活性天然产物相关研究报道,因此,极地微生物极具研究价值[17-18]。
笔者以一株采自北极海域海绵共附生放线菌Streptomyces sp. LHW11-07为研究对象,从其发酵浸膏中分离得到9个单体化合物1~9,包括环二肽化合物1~5,倍半萜化合物6,核苷类化合物7,以及两个其他结构类型化合物8和9,其中化合物1和2是首次分离于Streptomyces放线菌,而这些化合物的生物活性还有待进一步探究;本研究进一步丰富了该属放线菌的化学多样性,同时,为高值化开发利用极地微生物这一国家战略资源提供了物质基础和理论依据。
据文献报道,化合物1对所测试的病原性细菌和真菌均具有一定的对抗作用[19],化合物2测试了4种肿瘤细胞均无明显的细胞毒性[20],化合物3对海胆Strongylocentrotus intermedius胚胎具有细胞毒活性[9],化合物5具有抗炎活性并对H1N1和RSV病毒有一定的杀伤作用[21],化合物7作为一种内源性嘌呤核苷,具有降低血压、抑制血小板聚焦、舒张血管、减慢心律等生理活性[22],而化合物9可抑制癌细胞株的增殖,在体外对Trypanosoma brucei brucei GUTat 3.1表现出抗锥虫体活性等[23]。
-
表 1 多拉司琼DUE标准
评价指标 标准内容 预期目标值(%) 一级指标 二级指标 用药指征 适应证 适用于:①预防初次和重复使用致吐性肿瘤化疗(包括高剂量顺铂)引起的恶心和呕吐;②预防或治疗手术后恶心和/或呕吐。 100 禁忌证 已知对本品过敏的患者禁用。 100 用药过程 用法用量 ①预防和治疗化疗引起的恶心呕吐:1.8 mg/kg 或是固定剂量100 mg;②预防或治疗手术后恶心和/或呕吐:12.5 mg。 80 给药时间 ①预防和治疗化疗引起的恶心呕吐:化疗前30 min给药;②预防手术后恶心和/或呕吐;麻醉停止前15 min;③治疗手术后恶心和/或呕吐:刚出现恶心、呕吐时。 100 溶媒 预防和治疗化疗引起的恶心呕吐:溶于50 ml 溶媒(0.9%氯化钠注射液或是5%葡萄糖注射液)静脉输注15 min 以上。 80 特殊人群 ①儿童:尚无2岁以下患儿使用本品经验,2~17岁患儿耐受性好;
②妊娠期:尚缺乏在怀孕妇女中的充分的良好对照的研究;
③哺乳期:本品是否乳汁排泄尚不清楚,慎用;
④65岁以上患者不需要调整剂量,但用药仍须谨慎;
⑤肾功能不全:无需调整剂量;
⑥肝功能不全:无需调整剂量。100 疗程 化疗期间或是化疗后,存在恶心呕吐感即可。 100 联合用药 单药不能控制的恶心呕吐,可联合使用激素地塞米松,严重的可再联合NK-1抑制剂阿瑞吡坦。 100 相互作用 本品不能与其它药物混合使用,输注后要冲洗输液管道。 100 用药结果 不良反应 发生本品不良反应停用本品并对症处理,上报不良反应相关信息至ADR监测系统和药剂科。 100 疗效 恶心呕吐次数减少或消失。 90 管理指标 病程记录 ①病程录中有相关的用药指证;
②联合用药以及对患者恶心呕吐严重程度的描述。90  下载: 导出CSV
下载: 导出CSV
表 2 多拉司琼DUE评价结果
评价指标 标准符合率% 一级指标 二级指标 用药指征 适应证 96.9 禁忌证 100.0 用药过程 用法用量 43.3 给药时间 100.0 溶媒 23.7 特殊人群 100.0 疗程 100.0 联合用药 86.6 相互作用 100.0 用药结果 不良反应 100.0 疗效 100.0 管理指标 病程记录 68.0
下载: 导出CSV
-
[1] 周海辉, 张海霞, 葛卫红 化疗致恶心呕吐的研究进展[J]. 中国药师2018 21 7 1262 1265 .[2] 袁浩宇, 林勇, 胡明, 等 药物利用评价标准建立的方法探讨及实践[J]. 中国药房2010 21 22 2101 2104 .[3] 吕纹, 徐珺, 陈开盛,等 昂丹司琼临床应用的药物利用评价[J]. 药物流行病学杂志1995 4 4 230 232 .[4] 王东, 赵云燕, 朱虹, 等 某三甲医院司琼类药物应用情况与用药合理性分析[J]. 安徽医学2017 38 8 1001 1004 .[5] 王桂凤, 李雪芹, 刘锐锋, 等 某院替加环素药物利用评价标准的建立与应用分析[J]. 中国药房2017 28 14 1892 1895 .[6] 冯晓俊, 邓明影, 李宇, 等 华法林药物利用评价标准的构建及运用研究[J]. 安徽医药2018 22 11 2258 2262 .[7] ROCHFORD M, KIERNAN T J, AZIZ A Dolasetron overdose resulting in prolonged QTc interval and severe hypotension: a case report and literature review[J]. Emerg Med J2007 24 7 515 517 .[8] 高胜男, 刘国强 药物经济学在医药卫生领域的应用[J]. 中国药物经济学2017 12 8 16 18 .[9] DIMMITT D C, HUNT T L, SPALITTO A J, et al Effect of infusion rate on the pharmacokinetics and tolerance of intravenous dolasetron mesylate[J]. Ann Pharmacother1998 32 1 39 44 .[10] DIMMITT D C, SHAH A K, ARUMUGHAM T, et al Pharmacokinetics of oral and intravenous dolasetron mesylate in patients with renal impairment[J]. J Clin Pharmacol1998 38 9 798 806 . -

