


 下载:
下载:

-
胆管癌(cholangiocarcinoma, CCA)是胆道系统一组致死率极高的上皮细胞恶性肿瘤,按照解剖学定位,可分为肝内胆管癌(ICC)和肝外胆管癌(ECC), 后者又分为肝门部胆管癌(PCC)和远端胆管癌(DCC)[1]。胆管癌在原发性肝癌中占15%左右,仅次于肝细胞癌(HCC),我国胆管癌的发病率约为7.5/10万,而泰国东北部的发病率是我国的11倍[2-3]。全球胆管癌的死亡率为1~6/10万,并在全球范围内有上升之势,严重威胁着人民的生命健康[4]。
手术切除是唯一可能治愈胆管癌的方法,但因早期胆管癌症状不典型,绝大多数患者(60%~70%)在诊断时已经处于疾病中晚期,错过了肝病治愈性切除的最佳时机[5-6]。同时,胆管癌对放化疗不敏感且存在耐药现象,胆管癌的预后非常差[7]。胆管癌中位生存时间为24个月,5 年死亡率高达90%[8]。早期发现的胆管癌患者的生存率远高于晚期诊断的患者[9],因而,早期诊断胆管癌显得尤为重要。目前,胆管癌早期诊断普遍使用的是影像学检查以及肿瘤标志物临床筛查。超声、CT 和核磁共振成像等影像学方法是通过肝内占位来判断胆管癌,其准确性不佳,易出现漏诊和误诊,且对人体组织具有一定的伤害性[10]。癌胚抗原(CEA)和糖类抗原19-9(CA19-9)和125(CA125)是临床上用于胆管癌早期诊断和预后监测的生物标志物[11]。这些生物标志物的灵敏度和特异性较差,尚不能满足临床的需求[12]。因此,现在急迫地需要寻找和开发特定的生物标志物,用于胆管癌的早期诊断和预后监测。
在胆管癌的发生和发展过程中,患者机体内的糖类、氨基酸和脂质等内源性小分子代谢物会发生异常变化[4]。代谢物不仅是基因和蛋白质表达的最终产物,也是基因和内部环境相互作用的结果[13-14]。因此,代谢物浓度的变化有利于揭示机体疾病的病理生理过程。代谢组学已经发展成为一种技术工具,它使用高通量、高灵敏度的分析技术来筛选生物样品中与机体功能变化密切相关的低分子量代谢物[15-16]。代谢组学通过分析和验证疾病的特定生物标志物,在此基础上寻找相关代谢途径,使我们能够更好地了解胆管癌的病理过程和物质代谢途径,最终帮助胆管癌的临床诊疗。本文主要对代谢组学在胆管癌发病机制和复发机制、早期诊断、药理研究和药效评价中的研究进展进行综述。
-
生物体受到病理生理刺激或基因修饰时,可发生动态的多参数代谢反应,代谢组学就是定性和定量分析上述代谢反应过程中所有低分子量代谢物的科学[17-18]。代谢组学是一种新兴的技术工具,用来检测和分析来自体液、细胞以及组织等生物系统中分子质量小于1 000的各种代谢物,如糖类、脂类、氨基酸、有机酸、小分子肽以及维生素等[19]。代谢组学研究的一个关键步骤是在靶向代谢组学[20]和非靶向代谢组学[21]之间进行选择。前者寻求一组预定义的代谢物,可用于识别和验证特定代谢物,后者将在没有定义目标列表的情况下检测数百或数千种代谢物,不仅可以表征一般代谢特征的变化,还可以检测以前未知的代谢物[22-23]。
核磁共振(NMR)、液相色谱与质谱(LC-MS)联用[24]或气相色谱与质谱(GC-MS)联用[25]是代谢组学三个主要的化学分析平台。每一种分析平台都有自己的优缺点。NMR包括氢谱(1H-NMR)、碳谱(13C-NMR)以及二维谱(2D-NMR)等检测类型,能够对样本实现无创、非靶向的检测且重复性好,几乎不需要样品制备,可以分析质谱难以电离或需要衍生反应的代谢物,但灵敏度低、检测范围有限[19, 26]。在代谢组学研究中,质谱一般和液相色谱或气相色谱结合使用。经过色谱的前期分离作用,可以减少离子抑制效应,提高了质谱对代谢物定性和定量的灵敏度和分辨力。GC-MS一般需要先对样本进行衍生化后再分析,其优势在于灵敏度高、稳定性强以及有标准谱图库可快速鉴别代谢物 [27]。LC-MS不需要对样品进行复杂的衍生化处理,具有前处理简单、高分辨率、分离效率高、专属性强以及检测范围广的技术优势 [28]。多种分析技术进行联用,能够相互补充,发挥代谢组学高通量、高灵敏度以及高准确度的优势,更全面的寻找差异代谢物,这也是今后代谢组学分析技术发展的必然趋势。
-
代谢重编程被认为是癌症发展的一个标志,因为它需要平衡能量供给,以支持癌细胞的快速生长和繁殖[29]。在癌细胞中,即使在氧气充足、线粒体功能完整时,葡萄糖摄取率亦有明显的提高,且趋向糖酵解生成乳酸[30]。这就是1924年由奥托·首先注意到的Warburg效应。除此之外,其他代谢途径的改变,如脂肪生成和氨基酸代谢,也被报道与肿瘤的进展有关[31-32]。因此,侧重于分析生物样品中糖类、脂类、氨基酸以及有机酸等小分子代谢物的代谢组学,有利于揭示胆管癌发病和复发的机制。
研究表明,肝内胆管癌患者胆汁中磷脂酰胆碱含量降低,甘氨酸和牛磺酸结合胆汁酸含量升高,这与磷脂的胆汁排泄基因受损有关[33-34]。Murakami等[35]使用毛细管电泳-飞行时间质谱(CE-TOFMS)对手术切除后获得的10例肝内胆管癌样本和6例肝细胞癌样本进行转录组和代谢组分析,以探讨肝内胆管癌和肝细胞癌的发病机制。将样本分为肝内胆管癌、肝细胞癌、ICC-NT和HCC-NT四个样本组。主成分分析(PCA)发现,3-磷酸甘油、琥珀酸、甘油磷酰胆碱以及多种氨基酸在肿瘤组织的含量要低于非肿瘤组织。与其他三个样本组相比,肝内胆管癌样本组的次黄嘌呤和牛磺酸的含量显著增高。转录组分析结果表明,肝内胆管癌中mRNA的异常表达与异常的tRNA代谢、氨基酸代谢和脂蛋白代谢有关。转录组和代谢组的研究结果与此前的研究相一致。结合转录组和代谢组的通路分析发现,糖酵解、甘油磷脂代谢、氨基酸代谢以及氨酰-tRNA 生物合成与肝内胆管癌的致癌作用相关,说明糖脂代谢、氨基酸代谢、RNA代谢与肝内胆管癌的发生密切相关。
Padthaisong等[36]采用1H-NMR 和LC-MS分别对78例术后有无复发胆管癌患者(有40例,无38例)的血清进行了代谢组学和脂质组学研究。经秩和检验和正交偏最小二乘判别分析(OPLS-DA)等方法发现,与无复发患者相比,复发患者参与能量和氨基酸代谢的代谢物含量偏低,而大多数脂质TGs、PCs、PEs和PAs的含量升高。对所有显著差异代谢物进行了受试者工作特征曲线(ROC)分析,发现柠檬酸、肌氨酸、琥珀酸、肌酸、肌酸酐、丙酮酸以及TGs对胆管癌复发有良好的预测价值(P<0.05,AUC>0.7)。同时,复发患者的葡萄糖和丙酮酸水平较低,与糖酵解和丙酮酸代谢活性较高有关,柠檬酸和琥珀酸水平较低,与TCA循环活性较低有关。大多数的脂质升高,表明脂质在胆管癌的复发中发挥了很重要的作用。有研究表明,线粒体呼吸作用的低活性以及脂质摄取的诱导与癌症干细胞样细胞 (CSC)有关[37]。Padthaisong等[36]进一步研究发现,参与脂质摄取的蛋白质CD36,其高表达与CSC标记物相关。此外,高水平的CD36与较低的无复发生存率相关,这表明胆管癌复发的机制可能与CSCs有关。
-
Liang等[38]采用超高效液相色谱-四极杆/飞行时间串联质谱仪(UPLC-QTOF/MS)对肝细胞癌与肝内胆管癌患者的血浆进行了脂质组学研究。由于区分肝内胆管癌和肝细胞癌是诊断肝内胆管癌的一个难点,因此该研究通过分析109例健康志愿者、85例肝细胞癌患者与98例肝内胆管癌患者的样本,成功构建出区分肝细胞癌患者与肝内胆管癌患者的诊断模型。与肝细胞癌患者组相比,在肝内胆管癌组中鉴定出14种差异代谢物。进一步分析发现,与肝内胆管癌患者组相比,肝细胞癌患者组的磷脂酰乙醇胺[PE(19∶0/0∶0)]、磷脂酰胆碱[PC(14∶0/0∶0)]和[PC(18∶0/0∶0)]水平显著升高,而PE[18∶2(9Z,12Z)/0∶0]水平明显降低(VIP>1.40,P<0.01),它们之间的代谢差异主要发生在甘油磷脂的代谢中。经过进一步的工作特征曲线分析,PE(19∶0/0∶0)、PE[18∶2(9Z,12Z)/0∶0]、PC(14∶0/0∶0)和PC(18∶0/ 0∶0) 的曲线下面积(AUC)分别是0.918、0.833、0.807和 0.784,而结合这4种代谢物对区分肝细胞癌和肝内胆管癌患者的诊断准确率可达到 99.7%。这表明,作为代谢组学重要组成部分的脂质组学,可以用于肝细胞癌和肝内胆管癌的早期诊断与区分。
Wang等[39]应用超高效液相色谱-电喷雾离子化-四极杆/飞行时间串联质谱仪(UPLC-ESI-QTOF/MS)对60例肝外胆管癌患者和60例健康志愿者的尿液进行非靶向代谢组学分析,鉴定出19种差异代谢物,它们包括3-甲基组氨酸、柠檬酸、胞嘧啶、吲哚乙酸、水杨酸、L-蛋氨酸、氨基丙二酸、戊二酸、熊去氧胆酸、N-乙酰鸟氨酸、尿囊素、甘氨胆酸、组胺、尿黑酸、L-犬尿氨酸、肌氨酸、丙酮酸、牛磺酸和甲基琥珀酸(VIP>10,P<0.05)。其中11种差异代谢物含量增加,8种含量降低。代谢途径分析表明,差异代谢物的出现可能与肝外胆管癌患者紊乱的牛磺酸和亚牛磺酸代谢、丙酮酸代谢以及三羧酸循环有关。这19种生物标志物具有作为肝外胆管癌早期诊断的潜力。
Haznadar等[40]应用超高效液相色谱-串联质谱法(UPLC-MS/MS)对98例肝细胞癌患者、101例高危受试者和95例健康受试者尿液样本中的肌酸核苷(CR)、N-乙酰神经氨酸(NANA)、硫酸皮质醇和一种脂类分子(561+)进行定量分析,随后在117例肝细胞癌和253例肝内胆管癌患者,228例肝细胞癌和243例肝内胆管癌高危受试者,251例健康受试者的队列中进行了验证。并对肝内胆管癌和肝细胞癌患者组织样本中的CR和NANA进行定量分析。采用ANOVA方差分析、Wilcoxon 秩和检验和 COX回归分析等统计分析方法来评估各组4种代谢物的差异水平。结果发现,在两个队列中,肝细胞癌和肝内胆管癌患者的这4种代谢物均显著升高。进一步采用ROC分析发现,这4种代谢物比当前临床使用的标记物(CA19-9)能更好地区别肝内胆管癌和肝细胞癌患者。把它们结合起来,可得到一个显著优化的诊断模型,该模型AUC可达到0.88,灵敏度和特异性分别为71%和92%。同时,肝内胆管癌患者尿液和组织样本中CR和NANA含量较肝细胞癌患者有明显增加(P=0.002,P=0.02)。其中,高水平的CR与较差的肝内胆管癌预后相关。
Banales等[41]采用超高效液相色谱-飞行时间串联质谱仪(UPLC-TOF/MS)对健康志愿者和肝内胆管癌、肝细胞癌、原发性硬化胆管炎患者的血清进行了代谢组学研究。鉴定出的差异代谢物与CA19-9进行对比分析,其AUC均比CA19-9 的大。进一步应用广义线性模型(GLM)建立不同代谢物结合的诊断模型,并评估模型效能,发现将甘氨酸、天冬氨酸、鞘磷脂(42∶3)和鞘磷脂(43∶2)结合在一起能够准确的区分肝内胆管癌和肝细胞癌患者。该模型的AUC为0.890,灵敏度为75%,特异性为90%。另一种将磷脂酰胆碱(34∶3)和组氨酸相组合,可准确区分硬化胆管炎和肝内胆管癌,AUC为0.990,灵敏度为100%,特异性为70%。说明血清代谢物浓度的变化有望区分肝内胆管癌患者和健康人群, 不同代谢物的组合有助于肝内胆管癌与肝细胞癌或硬化胆管炎的早期鉴别诊断。
Macias等[42]通过LC-MS技术对远端胆管癌患者、胰导管腺癌(PDAC)患者以及良性胰腺疾病(BPD)患者的血清进行了代谢组学研究,并成功构建了能够用于远端胆管癌与胰导管腺癌早期诊断的模型:结合酰基肉碱(16∶0)、神经酰胺(D18∶1/24∶0)、磷脂酰胆碱(20∶0/0∶0)和(O-16∶0/20∶3)、溶血磷脂酰胆碱(20∶0/0∶0)和(0∶0/20∶0)、溶血磷脂酰乙醇胺(P18∶2/0∶0)、鞘磷脂(D18∶2/22∶0)和(D18∶2/23∶0) 9种代谢物的模型,能够区分远端胆管癌与胰导管腺癌患者。该模型的AUC为0.85,灵敏度为55.9%,特异性为89.5%。将这9个代谢物与CA19-9结合可以进一步提高诊断模型的灵敏度,模型的AUC为0.888,灵敏度为71.4%,特异性为89.2%。这表明获得的差异代谢物具有作为生物标志物的潜力,他们的结合可将远端胆管癌患者与胰导管腺癌患者区分,应用于早期诊断。
-
Kotawong等[43]采用LC-MS/MS,对暴露于苍术素后的胆管癌细胞提取物进行代谢组学研究。发现在使用苍术素后,柠檬酸、琥珀酸含量显著升高,谷胱甘肽二硫化物(GSSG)含量显著下降。其中GSSG随时间的增加,下降趋势会更明显。柠檬酸和琥珀酸含量升高,说明苍术素能促进三羧酸循环。GSSG是氧化型谷胱甘肽,与还原型谷胱甘肽(GSH)相互转化。在正常生理状态下,GSH和GSSG维持动态平衡,而GSH的降低、GSSG水平的升高是导致癌症的重要因素[44]。因此,苍术素可能通过促进三羧酸循环,下调氧化型谷胱甘肽的水平,而发挥抗胆管癌作用。
Zhang等[45]采用NMR对胆管癌细胞提取的代谢物进行了代谢组学研究。结果发现二甲双胍干预后,胆管癌细胞中与能量代谢相关的丙酮酸、乳酸、NAD+以及多种必需氨基酸含量升高,而葡萄糖、三磷酸鸟苷、α-酮戊二酸、延胡索酸以及大多数非必需氨基酸含量降低。进一步探讨二甲双胍干预的胆管癌细胞和正常人脐静脉内皮细胞 (HUVEC)的代谢差异,发现使用二甲双胍后,胆管癌细胞中葡萄糖和延胡索酸含量降低,NAD+含量升高,而HUVEC的代谢变化相反。造成这种代谢差异的原因是癌细胞的代谢重编程(Warburg效应),无论氧气水平如何,癌细胞都会发生高速率的糖酵解和乳酸发酵,二甲双胍的干预对糖酵解有促进作用,并将NADH的电子排出,使丙酮酸变成乳酸,因而使癌细胞的Warburg效应增强。糖酵解的调节与AMPK高度相关,而二甲双胍可激活AMPK[46-47]。这些与糖酵解相关的代谢物变化为胆管癌药效机制研究提供了一个新视角。
Chen等[48]采用UPLC-QTOF/MS对肝内胆管癌小鼠及蛇床子素给药后小鼠血清进行代谢组学研究。与对照组小鼠相比,模型组小鼠主要表现为氨基酸和脂质代谢失调。在AKT/YapS127A共表达诱导的肝内胆管癌小鼠血清中,鸟氨酸、组氨酸、色氨酸、蛋氨酸和泛酸等11种代谢物含量升高,肉碱和溶血磷脂酰胆碱[16∶1(9Z)/0∶0]含量降低,在脂质代谢方面,肝内胆管癌组的大部分血脂水平较高。泛酸与脂肪酸的合成有关,肉碱水平的降低,是因为癌细胞中脂肪酸代谢的破坏和脂肪酸的 β-氧化减少。在蛇床子素给药后,可以逆转这些潜在生物标志物的变化趋势,使它们的水平逐渐恢复正常。说明蛇床子素的药效机制可能与改善肝内胆管癌紊乱的氨基酸代谢、脂肪酸代谢以及甘油磷脂代谢相关。
-
综上所述,作为系统生物学研究的重要组成部分,代谢组学寻找胆管癌潜在的生物标志物,为胆管癌的机制研究以及疾病的早期诊断、指导治疗决策和个体化靶向治疗等提供了新的视角。相对于其他组学而言,代谢组学具有操作简单、创伤小、所需样本量少和重复性强等优势。胆管癌是一种与代谢密切相关的恶性肿瘤,目前主要通过组织、血清和尿液等样本进行代谢组学研究,寻找差异代谢物构建早期诊断模型以及探讨疾病发病机制。同时,关键通路相关的代谢物具有作为胆管癌新治疗靶点的潜力。代谢组学对阐明胆管癌的发病机制、早期诊断、监测复发及预后判断具有较好的前景。然而,虽然现有的研究已经发现了许多与胆管癌相关的差异代谢物,但是如何阐述各个潜在的生物标志物之间的关联与其背后深层次原因仍旧是一个难点和挑战。部分研究样本量较少,缺乏交叉验证,这使实验结果的准确性受到一定的影响。同时,代谢是生物体内的一个动态变化的过程,受到多种理化因素的影响,代谢组学提供给系统生物学的研究信息仅限于代谢物,无法表征完整的表型,它不足以全面解决多种复杂的生物学问题。因此,将多个组学领域(基因组学、蛋白质组学和代谢组学)、多种分析技术(LC-MS、GC-MS和NMR)以及不同样本类型(组织、血清和尿液)整合起来,以加强对疾病生物学机制和生物标志物相关性的理解,是未来代谢组学发展的一个方向。
Advances in metabolomics of cholangiocarcinoma
-
摘要: 胆管癌(CCA)恶性程度高,由于早期缺乏典型症状且尚无准确的生物标志物,发现时常处于晚期,预后较差。胆管癌的发生发展与代谢密切相关,代谢组学是研究在病理生理或基因修饰等因素影响下,生物体内内源性小分子代谢物变化的学科,具有全局分析、高通量和反映生物体系实时变化的特点,可以为胆管癌生物标志物的筛选、疾病诊疗提供新途径。综述近年来代谢组学在胆管癌方面的研究进展,以期为进一步研究提供参考。Abstract: As a highly malignant tumor, the diagnosis of cholangiocarcinoma (CCA) is often late and the prognosis is poor for which the early symptoms are atypical and the lack of accurate biomarkers. Metabolomics is an emerging science that researches the alterations of all endogenous small molecule metabolites in an organism under the influence of pathological, physiological or genetic modification. The development and progress of CCA is closely related to metabolism. Metabolomic is characterized by global analysis, high throughput and reflects real-time alterations in biology system, providing a new avenue for biomarker screening and diseases diagnosis and treatment. The advances of metabolomics studies on CCA in the recent years were reviewed in this paper which could provide the reference for further research.
-
Key words:
- cholangiocarcinoma /
- metabolomic /
- biomarkers /
- mechanism /
- early diagnosis /
- pharmacodynamic evaluation
-
胰腺癌是一种病死率极高的消化道恶性肿瘤 [1]。超过80%以上的患者一旦确诊即是晚期,手术难以根治,需在术后进行辅助化疗、放疗、对症支持治疗等 [2]。盐酸吉西他滨(hcGEM)是治疗胰腺癌的一线化疗药。由于存在半衰期短、产生耐药性及不可避免的毒副作用等问题,其疗效不尽如人意[3]。因此,hcGEM的临床应用需要联合化疗来提高疗效[4]。
铁死亡是一种铁依赖的非凋亡性细胞死亡形式,针对铁死亡的治疗策略可能为传统疗法难以攻克的癌症提供新的治疗思路 [5]。埃拉斯汀 (Era)作为一种高效持久的铁死亡诱导剂,它可以激活多种信号通路来触发癌细胞的铁死亡 [6]。柳氮磺胺吡啶(SASP)是一种能抑制铁死亡相关的胱氨酸-谷氨酸逆向转运蛋白的抗炎药,可通过降低癌细胞对胱氨酸的摄取以及胞内谷胱甘肽水平来抑制胰腺癌细胞的生长[7]。青蒿琥酯(Art)是一种青蒿素的衍生物,除用作抗疟治疗外,可通过促进铁蛋白吞噬来增加细胞内游离铁水平,进而引发癌细胞的铁死亡[8]。铁死亡诱导剂与盐酸吉西他滨联合应用可能是胰腺癌治疗的潜在策略[9]。
本研究分别考察Era、SASP和Art这三种铁死亡诱导剂单独或联合hcGEM使用,对人胰腺癌PANC-1细胞的增殖抑制作用,以期发现具有潜在协同抑制的联合方案,为今后开发胰腺癌新疗法奠定基础。
1. 实验材料
1.1 试剂
盐酸吉西他滨、柳氮磺胺吡啶、埃拉斯汀(美国MCE公司);青蒿琥酯(上海泰坦科技股份有限公司);胎牛血清、青链霉素、胰酶(以色列BI公司);DMEM高糖培养基、PBS缓冲液(上海泰坦科技股份有限公司),CCK- 8细胞毒性试剂盒(日本同仁化学研究所)。
1.2 细胞株
人胰腺癌PANC-1细胞,来源于海军军医大学第一附属医院消化内科,冻存复苏后培养于含10%胎牛血清的DMEM高糖培养基。
2. 实验方法
2.1 细胞培养
人胰腺癌PANC-1细胞用DMEM完全培养基(含10 %胎牛血清,1 %青-链霉素)于5 % CO2、37 ℃的恒温培养箱中培养。待细胞融合度达80 %~90 %时,移除旧培养基,PBS缓冲液清洗2遍后加入适当体积的胰蛋白酶消化3 min。待大部分细胞镜下变圆,小部分细胞脱落时,加入2倍胰酶体积的完全培养基终止消化,充分吹打使细胞脱离。将细胞悬液以1 000 r/min转速,离心5 min,弃上清液,加入2 ml完全培养基重悬,并按1∶2的比例均匀接种于两个培养皿,加入8 ml完全培养基水平摇匀后置于恒温细胞培养箱内培养,取对数生长期的细胞进行后续细胞毒性实验。
2.2 实验分组
将实验细胞单独用药组根据药物种类与浓度不同,分为hcGEM单药组(hcGEM: 0.015 625、0.031 25、0.062 5、0.125、0.25、0.5、1、2、4 μmol/L),Era单药组(Era:0.25、0.5、1、2、4、8、16、32、64 μmol/L),SASP单药组(SASP: 6.25、12.5、25、50、100、200、400、800、1600 μmol/L)以及Art单药组(Art:0.5、1、2、4、8、16、32、64、128 μmol/L),每种药物都按2倍比率设置浓度梯度,每组各9个浓度;联合用药组根据联合药物组成与比例不同分为hcGEM-Era联合用药组(包括4∶1、1∶1、1∶4和1∶16 联合组)、hcGEM-SASP联合用药组(包括4∶1、1∶1、1∶20和1∶400联合组)、hcGEM-Art联合用药组(包括 2∶1、1∶1、1∶4和1∶16联合组)。每个联合用药组中,hcGEM的浓度均为相同的浓度梯度(hcGEM: 0.015 625、0.031 25、0.062 5、0.125、0.25、0.5、1、2、4 μmol/L),并与恒定比例的联合药物共同作用于细胞。hcGEM-Era 1∶16联合组的药物浓度梯度为0.015 625 μmol/L hcGEM+0.25 μmol/L Era、0.031 25 μmol/L hcGEM+0.5 μmol/L Era、0.062 5 μmol/L hcGEM+1 μmol/L Era、0.125 μmol/L hcGEM+2 μmol/L Era、0.25 μmol/L hcGEM+4 μmol/L Era、0.5 μmol/L hcGEM+8 μmol/L Era、1 μmol/L hcGEM+16 μmol/L Era、2 μmol/L hcGEM+32 μmol/L Era、4 μmol/L hcGEM+64 μmol/L Era。
2.3 CCK-8法检测细胞抑制情况
取对数生长的PANC-1细胞接种于96孔板,密度为5 000个细胞/孔。待细胞贴壁24 h后,弃上清液,每3个复孔加入100 μl不同浓度的每种药物,另设空白对照组与药物未处理组各3个复孔。药物与细胞共培养48 h后,每孔加入含10% CCK-8的DMEM培养基。于恒温培养2 h后,用酶标仪测量450 nm波长下各孔的吸光度值(A值)。使用GraphPad Prism 8.0.2软件对数据进行非线性回归分析,得到各组药物作用于细胞的半数抑制浓度(IC50) 以及细胞增殖抑制率,具体公式为:抑制率=[1−(A用药组−A空白组)/(A未用药组−A空白组)]×100%。
2.4 联合指数计算
根据单独用药组与联合用药组的摩尔浓度与细胞增殖抑制率,使用CompuSyn软件计算联合指数(CI),并依据CI值判断药物的联合效果: CI>1表示拮抗作用;CI=1表示相加作用;CI<1表示协同作用,且协同抑制效果随CI值的减小而增强。
2.5 统计学分析
实验结果以(
$\bar x $ ±s)表示,采用GraphPad Prism 8.0.2软件进行数据的处理分析,多组间比较采用单因素方差分析,两组间比较采用t检验。P<0.05表示组间差异有统计学意义。3. 结果
3.1 hcGEM、Era、SASP、Art单独用药对PANC-1细胞的影响
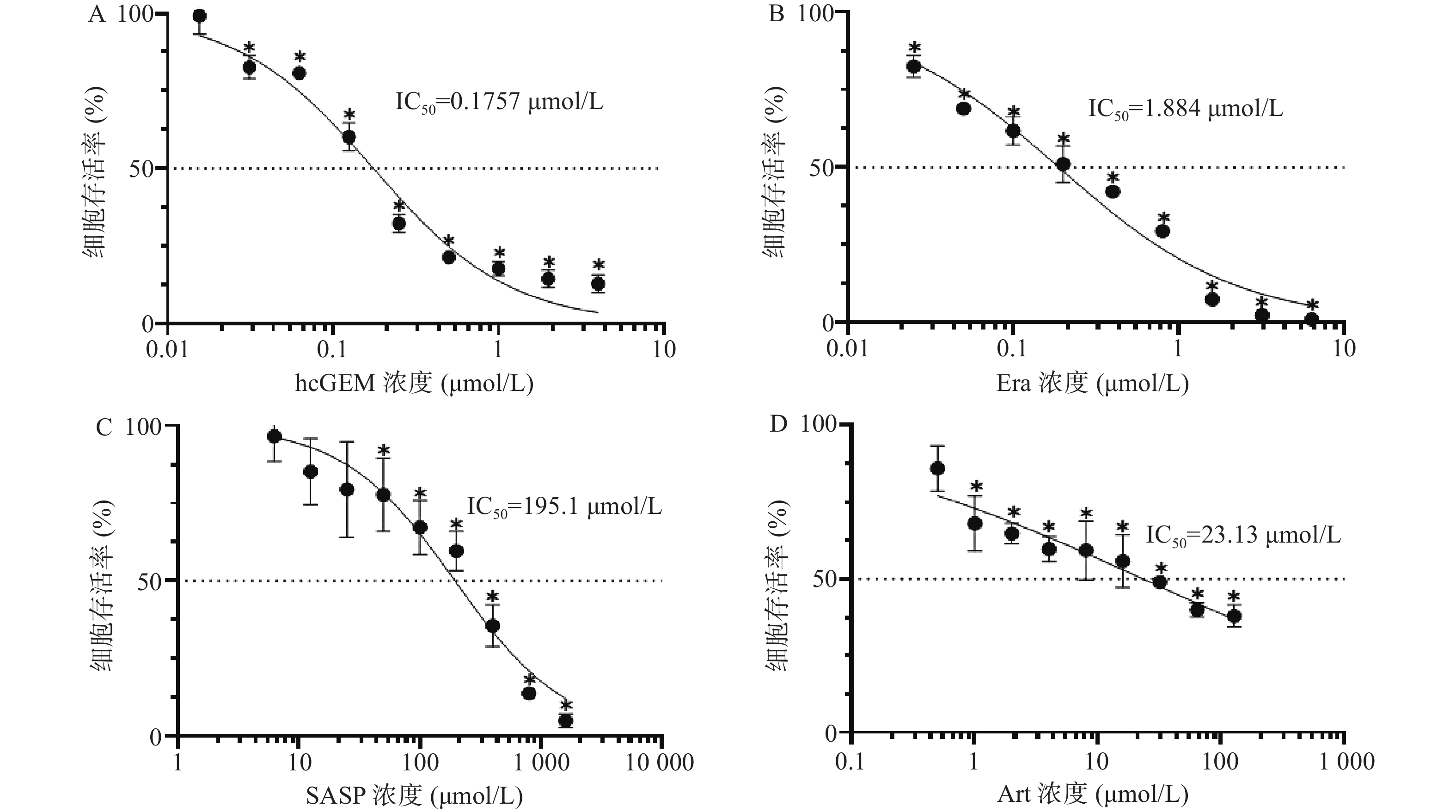
如图1所示,hcGEM、Era、SASP、Art单独作用于PANC-1细胞时可明显抑制细胞的增殖,并且这种抑制作用呈现剂量依赖性。hcGEM、Era、SASP、Art作用于PANC-1细胞的IC50值分别为0.175 7、1.884、195.1、23.13 μmol/L。hcGEM单独用药时,小于0.015 625 μmol/L的剂量对PANC-1细胞的生长几乎无抑制作用,存活率>95%(P>0.05);剂量大于2 μmol/L时,细胞的活性受到显著抑制,存活率<15%(P<0.05)。Era单独用药时,大于16 μmol/L 的剂量能显著抑制PANC-1细胞的生长,存活率<10%(P<0.05),而小于0.25 μmol/L的剂量会使80%以上的细胞存活。SASP单独用药时,大于800 μmol/L的剂量能抑制PANC-1细胞的生长,存活率<15%(P<0.05),而小于12.5 μmol/L的剂量仅轻微抑制PANC-1细胞存活,存活率>85%(P>0.05)。Art单独用药时,大于128 μmol/L的剂量能显著抑制PANC-1细胞的生长,存活率<40%(P<0.05),而小于0.5 μmol/L时,仅能轻微抑制细胞增殖,存活率>85%(P>0.05)。
3.2 hcGEM分别与Era、SASP、Art联合用药对PANC-1细胞的影响
在研究单药对PANC-1细胞抑制效果的基础上,我们进一步探究了hcGEM-Era 4∶1、1∶1、1∶4和1∶16 联合用药,hcGEM-SASP 4∶1、1∶1、1∶20和1∶400联合用药,以及hcGEM-Art 2∶1、1∶1、1∶4和1∶16联合用药,对PANC-1细胞的抑制效果。如图2所示, hcGEM-Era、hcGEM-SASP以及hcGEM-Art联合用药组的抑制效果随着浓度的增加而提高。其中,hcGEM-Era 4∶1、1∶4、1∶16联合组,以及hcGEM-SASP 1∶400联合组对PANC-1细胞抑制效果在所有浓度梯度范围均优于hcGEM组(P<0.05)。此外,hcGEM-Era 4∶1、1∶4、1∶16联合用药组,hcGEM-SASP 1∶20、1∶400联合组,和hcGEM-Art 1∶16联合组的IC50值均小于0.1757 μmol/L(hcGEM单药组的IC50),说明上述联合用药能在一定程度上提高对PANC-1细胞的抑制效果。不同联合用药组hcGEM的IC50值见表1。
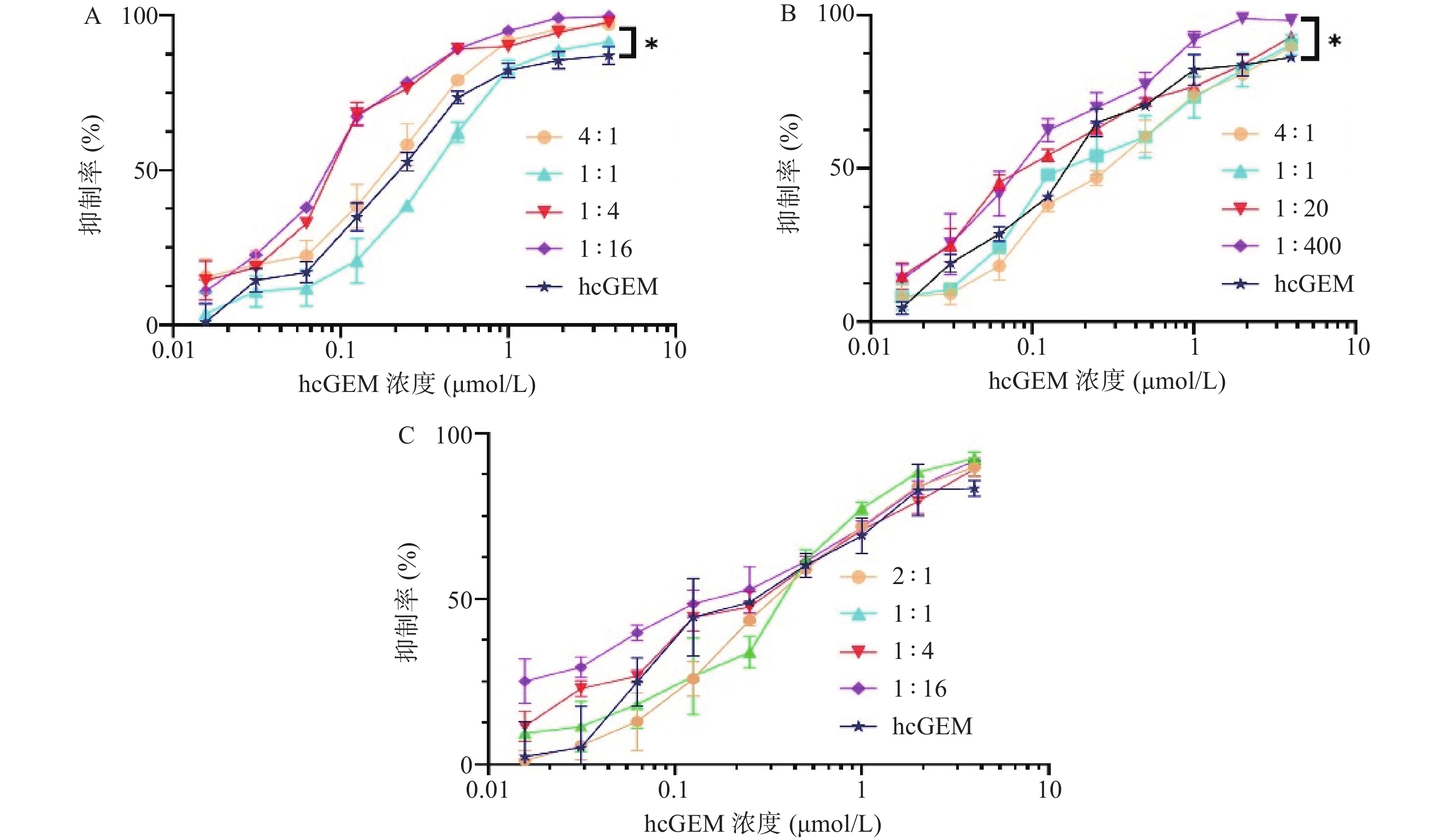 图 2 三种铁死亡诱导剂分别联用hcGEM对胰腺癌PANC-1细胞存活率的影响A.hcGEM-Era联合用药组;B.hcGEM-SASP联合用药组;C.hcGEM-Art联合用药组;*P<0.05, 与单用hcGEM组比较表 1 hcGEM联合三种铁死亡诱导剂对胰腺癌PANC-1细胞作用时的IC50值
图 2 三种铁死亡诱导剂分别联用hcGEM对胰腺癌PANC-1细胞存活率的影响A.hcGEM-Era联合用药组;B.hcGEM-SASP联合用药组;C.hcGEM-Art联合用药组;*P<0.05, 与单用hcGEM组比较表 1 hcGEM联合三种铁死亡诱导剂对胰腺癌PANC-1细胞作用时的IC50值组别 摩尔浓度比 IC50(μmol/L,hcGEM) hcGEM-Era联合组 1∶0.25 0.140 3±0.009 1 1∶1 0.340 2±0.018 3 1∶4 0.091 3±0.005 1 1∶16 0.083 3±0.002 5 hcGEM-SASP联合组 1∶0.25 0.297 5±0.016 1 1∶1 0.240 2±0.021 2 1∶20 0.120 8±0.008 9 1∶400 0.092 6±0.006 7 hcGEM-Art联合组 1∶0.5 0.366 4±0.018 7 1∶1 0.344 4±0.026 3 1∶4 0.249 3±0.015 7 1∶16 0.154 6±0.013 5 3.3 药物联合指数
为了研究hcGEM分别与Era、SASP、Art联合用药,对PANC-1细胞是否具有协同抑制作用,我们分别设计了hcGEM与三种铁死亡诱导剂的4种不同比例的联用方案,来探讨不同联合用药组对PANC-1细胞的协同抑制效果。CompuSyn软件分析结果显示,hcGEM-Era 4∶1或1∶4联用组在所有药物浓度下均能对PANC-1细胞产生良好的协同抑制效果(CI<1),且4∶1联用组的协同抑制效果略优于1∶4联用组。对于hcGEM-Era 1∶16联用组,CI值随着联合药物浓度的增加而减小,说明该比例下两药协同抑制效果随着浓度增大有所增强,而hcGEM-Era 1∶1联用组的CI值,仅除1 μmol/LhcGEM+1 μmol/L Era、0.031 25 μmol/L hcGEM+0.031 25 μmol/L Era联用组的CI<1外,其余浓度组CI>1,说明hcGEM-Era 1∶1联用组对PANC-1细胞几乎无协同抑制效果(图3A)。
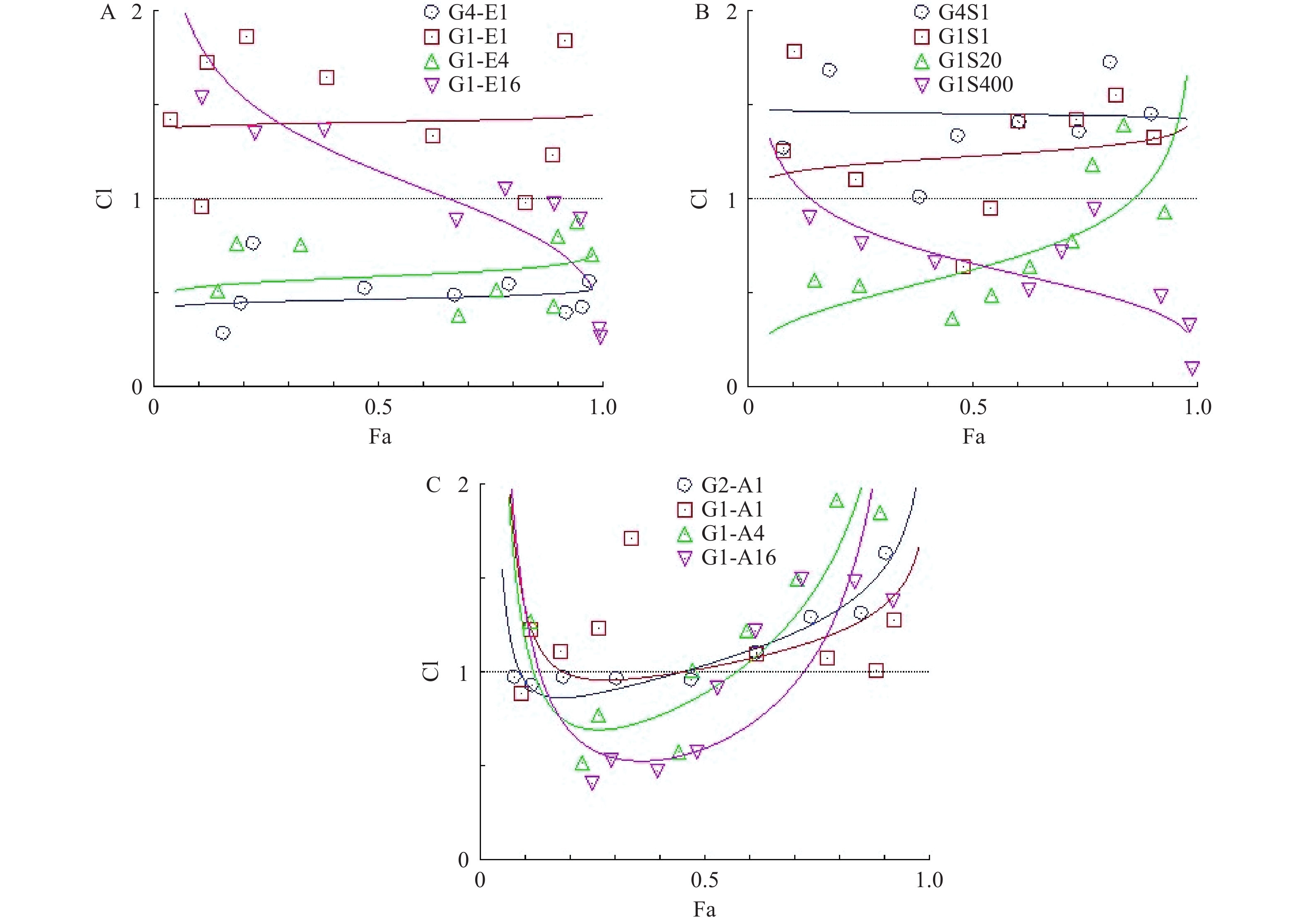 图 3 三种铁死亡诱导剂分别联用hcGEM对胰腺癌PANC-1细胞的效应-联合指数曲线图A.hcGEM-Era联用组;B. hcGEM-SASP联用组;C.hcGEM-Art联用组
图 3 三种铁死亡诱导剂分别联用hcGEM对胰腺癌PANC-1细胞的效应-联合指数曲线图A.hcGEM-Era联用组;B. hcGEM-SASP联用组;C.hcGEM-Art联用组对于hcGEM与SASP联合用药组对PANC-1细胞的协同抑制效果,CompuSyn分析结果提示,hcGEM-SASP 4∶1联用对PANC-1细胞没有协同作用(CI>1),而hcGEM-SASP 1∶1联用组除了0.25 μmol/L hcGEM+0.25 μmol/L SASP、0.125 μmol/L hcGEM+0.125 μmol/LSASP联用组CI<1外,其余联用浓度组的CI>1,表明hcGEM-SASP 1∶1联合用药对PANC-1细胞几乎无协同抑制作用。hcGEM-SASP 1∶20联合用药对PANC-1细胞,除了1 μmol/LhcGEM+20 μmol/L SASP、2 μmol/L hcGEM+40 μmol/LSASP联用组CI>1外,其余浓度组CI<1,提示hcGEM-SASP 1∶20联合用药对PANC-1细胞主要是协同抑制效果,且协同效果随着浓度的增加而减弱。hcGEM-SASP 1∶400联合用药对PANC-1细胞有良好的协同抑制效果(CI<1),且协同效果随着浓度的增加而增强(图3B)。
对于hcGEM与Art联合用药组对PANC-1细胞的协同抑制效果,CompuSyn结果提示,hcGEM-Art 2∶1联用组在0.031 25 ~0.25 μmol/L hcGEM联合浓度范围内CI <1,且接近1,其余联合浓度组的CI>1,表明hcGEM-Art 2∶1联合用药对PANC-1细胞几乎无协同抑制作用。hcGEM-Art 1∶1联用组CI>1,表明hcGEM-Art 1∶1联合用药对PANC-1细胞无协同抑制作用。hcGEM-Art 1∶4联用组仅在0.031 25~0.125 μmol/L hcGEM联合浓度范围内CI<1,其余联合浓度范围内CI>1,表明hcGEM-Art 1∶4联合用药对PANC-1细胞仅在0.031 25~0.125 μmol/L hcGEM联合浓度范围内存在协同抑制作用。 hcGEM-Art 1∶16联用组在0.015 625~0.25 μmol/L hcGEM联合浓度范围内CI <1,其余联合浓度范围内CI>1,表明hcGEM-Art 1∶16联用组在0.015 625~0.25 μmol/L hcGEM联合浓度范围内对PANC-1细胞有协同抑制作用 (图3C)。
4. 讨论
目前,胰腺癌的一线治疗标准为吉西他滨联合白蛋白结合型紫杉醇或者FOLFIRONOX组合(5-氟尿嘧啶、亚叶酸钙、伊立替康和奥沙利铂)[10]。吉西他滨在一定程度上能提高患者的生存率,但耐药性的出现以及毒副作用限制了临床治疗效果,故临床上常将其与其他化疗药联合使用来提高其疗效[11]。近来大量研究证实,诱导癌细胞铁死亡可能对包括胰腺癌在内的多种类型癌症有效[12]。自2003年发现小分子铁死亡诱导剂埃拉斯汀至今,研究人员已发现多种铁死亡诱导剂,如RSL3、Sorafeni、SASP、Art等[13]。本研究选取了三种铁死亡诱导剂Era、SASP以及Art,首先研究了三种铁死亡诱导剂单独应用对PANC-1细胞的增殖抑制作用。通过CCK-8法检测了三种铁死亡诱导剂的细胞毒性作用,我们发现Era对PANC-1细胞的IC50最小,而SASP的IC50最大,表明在这三种铁死亡诱导剂中,Era对PANC-1细胞的抑制作用最强,而SASP最弱。三种铁死亡诱导剂单独使用对PANC-1细胞的增殖抑制作用呈现剂量依赖性,即浓度越大抑制效果越强。
为了进一步探究三种铁死亡诱导剂Era、SASP、Art与hcGEM联用是否对胰腺癌PANC-1细胞具有协同抑制作用,本研究设计了不同比例的联合用药组来探索最佳的联合协同方案。本次研究结果显示,当三种铁死亡诱导剂分别与hcGEM联合使用时, hcGEM-Era 4∶1或1∶4联用组,hcGEM-SASP 1∶400联用组对PANC-1细胞具有良好的协同抑制效果。hcGEM-Art 1∶4或1∶16联用组仅在一定浓度范围内对PANC-1细胞有协同抑制效果。胰腺导管腺癌患者中,90%存在KRAS突变, 而致癌KRAS将胰腺导管腺癌细胞重新编程为高度抗凋亡的状态。由于KRAS信号突变的副产物是生成大量的ROS,为了上调抗氧化能力,胰腺导管腺癌转而增强葡萄糖和谷氨酰胺代谢途径,使细胞对ROS诱导的、铁依赖性非凋亡的铁死亡模式敏感[13]。激活铁死亡可有效阻止肿瘤进展,增强化疗、放疗和免疫治疗的效果[14]。在治疗胰腺癌的过程中,吉西他滨通过NF-κB信号途径诱导内源性活性氧的产生,进而导致Nrf2信号通路的激活以及细胞内谷胱甘肽增加,最终使胰腺癌细胞对吉西他滨不敏感[15]。埃拉斯汀可通过抑制胱氨酸-谷氨酸逆向转运蛋白的活性来减少胱氨酸进入肿瘤细胞,从而降低胞内谷胱甘肽的合成,抑制线粒体的电压依赖性阴离子通道等多种途径促进线粒体代谢紊乱、活性氧类物质以及脂质过氧化物的大量积累,增强吉西他滨对胰腺癌细胞的杀伤效果[16]。柳氮磺胺吡啶可通过抑制胱氨酸-谷氨酸逆向转运蛋白的功能促进胰腺癌细胞的铁死亡,逆转癌细胞的耐药性 [17]。青蒿琥酯通过诱导铁依赖性氧化损伤促进胰腺癌细胞的死亡,并且这种脂质过氧化性细胞死亡可被铁死亡抑制剂阻断[18]。本研究中三种铁死亡诱导剂联合hcGEM对PANC-1细胞的协同抑制的效果可能与这三种铁死亡诱导剂引起的氧化应激反应的强弱有关,具体相关机制还有待进一步实验。
综上所述,本研究发现三种铁死亡诱导剂分别与hcGEM联合应用时存在对PANC-1细胞的协同抑制联合方案。hcGEM-Era 4∶1或1∶4联合用药以及hcGEM-SASP 1∶400联合用药对PANC-1细胞的协同抑制效果良好,而hcGEM-Art 1∶4或1∶16联合用药仅一定浓度范围能对PANC-1细胞产生协同抑制效果。
-
[1] BRINDLEY P J, BACHINI M, ILYAS S I, et al. Cholangiocarcinoma[J]. Nat Rev Dis Primers,2021,7(1):65-81. doi: 10.1038/s41572-021-00300-2 [2] BANALES J M, MARIN J J G, LAMARCA A, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management[J]. Nat Rev Gastroenterol Hepatol,2020,17(9):557-588. doi: 10.1038/s41575-020-0310-z [3] BANALES J M, CARDINALE V, CARPINO G, et al. Expert consensus document: Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA)[J]. Nat Rev Gastroenterol Hepatol,2016,13(5):261-280. doi: 10.1038/nrgastro.2016.51 [4] RAGGI C, TADDEI M L, RAE C, et al. Metabolic reprogramming in cholangiocarcinoma[J]. J Hepatol,2022,77(3):849-864. doi: 10.1016/j.jhep.2022.04.038 [5] MASSIRONI S, PILLA L, ELVEVI A, et al. New and emerging systemic therapeutic options for advanced cholangiocarcinoma[J]. Cells,2020,9(3):688-709. doi: 10.3390/cells9030688 [6] VALLEJO A, ERICE O, ENTRIALGO-CADIERNO R, et al. FOSL1 promotes cholangiocarcinoma via transcriptional effectors that could be therapeutically targeted[J]. J Hepatol,2021,75(2):363-376. doi: 10.1016/j.jhep.2021.03.028 [7] RIZVI S, KHAN S A, HALLEMEIER C L, et al. Cholangiocarcinoma - evolving concepts and therapeutic strategies[J]. Nat Rev Clin Oncol,2018,15(2):95-111. doi: 10.1038/nrclinonc.2017.157 [8] DIGGS L P, RUF B, MA C, et al. CD40-mediated immune cell activation enhances response to anti-PD-1 in murine intrahepatic cholangiocarcinoma[J]. J Hepatol,2021,74(5):1145-1154. doi: 10.1016/j.jhep.2020.11.037 [9] BERTUCCIO P, MALVEZZI M, CARIOLI G, et al. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma[J]. J Hepatol,2019,71(1):104-114. doi: 10.1016/j.jhep.2019.03.013 [10] JOO I, LEE J M, YOON J H. Imaging diagnosis of intrahepatic and perihilar cholangiocarcinoma: recent advances and challenges[J]. Radiology,2018,288(1):7-13. doi: 10.1148/radiol.2018171187 [11] MUNUGALA N, MAITHEL S K, SHROFF R T. Novel biomarkers and the future of targeted therapies in cholangiocarcinoma: a narrative review[J]. Hepatobiliary Surg Nutr,2022,11(2):253-266. doi: 10.21037/hbsn-20-475 [12] RODRIGUES P M, VOGEL A, ARRESE M, et al. Next-generation biomarkers for cholangiocarcinoma[J]. Cancers (Basel),2021,13(13):3222-3246. doi: 10.3390/cancers13133222 [13] CHO K, MAHIEU N G, JOHNSON S L, et al. After the feature presentation: technologies bridging untargeted metabolomics and biology[J]. Curr Opin Biotechnol,2014,28:143-148. doi: 10.1016/j.copbio.2014.04.006 [14] GAUGUIER D. Application of quantitative metabolomics in systems genetics in rodent models of complex phenotypes[J]. Arch Biochem Biophys,2016,589:158-167. doi: 10.1016/j.abb.2015.09.016 [15] CHUA E C, SHUI G H, LEE I T, et al. Extensive diversity in circadian regulation of plasma lipids and evidence for different circadian metabolic phenotypes in humans[J]. Proc Natl Acad Sci U S A,2013,110(35):14468-14473. doi: 10.1073/pnas.1222647110 [16] SUHRE K, SHIN S Y, PETERSEN A K, et al. Human metabolic individuality in biomedical and pharmaceutical research[J]. Nature,2011,477(7362):54-60. doi: 10.1038/nature10354 [17] NICHOLSON J K, LINDON J C, HOLMES E. ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data[J]. Xenobiotica,1999,29(11):1181-1189. doi: 10.1080/004982599238047 [18] BINO R J, HALL R D, FIEHN O, et al. Potential of metabolomics as a functional genomics tool[J]. Trends Plant Sci, 2004, 9(9): 418-425. [19] GUMA M, TIZIANI S, FIRESTEIN G S. Metabolomics in rheumatic diseases: desperately seeking biomarkers[J]. Nat Rev Rheumatol,2016,12(5):269-281. doi: 10.1038/nrrheum.2016.1 [20] GRIFFITHS W J, KOAL T, WANG Y Q, et al. Targeted metabolomics for biomarker discovery[J]. Angew Chem Int Ed Engl,2010,49(32):5426-5445. doi: 10.1002/anie.200905579 [21] DENERY J R, NUNES A A, DICKERSON T J. Characterization of differences between blood sample matrices in untargeted metabolomics[J]. Anal Chem,2011,83(3):1040-1047. doi: 10.1021/ac102806p [22] DETTMER K, ARONOV P A, HAMMOCK B D. Mass spectrometry-based metabolomics[J]. Mass Spectrom Rev,2007,26(1):51-78. doi: 10.1002/mas.20108 [23] FIEHN O. Metabolomics: the link between genotypes and phenotypes[J]. Plant Mol Biol,2002,48(1-2):155-171. [24] CLIMACO PINTO R, KARAMAN I, LEWIS M R, et al. Finding correspondence between metabolomic features in untargeted liquid chromatography-mass spectrometry metabolomics datasets[J]. Anal Chem,2022,94(14):5493-5503. doi: 10.1021/acs.analchem.1c03592 [25] KISELEVA O, KURBATOV I, ILGISONIS E, et al. Defining blood plasma and serum metabolome by GC-MS[J]. Metabolites,2021,12(1):15-45. doi: 10.3390/metabo12010015 [26] MARKLEY J L, BRÜSCHWEILER R, EDISON A S, et al. The future of NMR-based metabolomics[J]. Curr Opin Biotechnol,2017,43:34-40. [27] JIN Q, MA R C W. Metabolomics in diabetes and diabetic complications: Insights from epidemiological studies[J]. Cells,2021,10(11):2832-2869. [28] LINDON J C, NICHOLSON J K. Spectroscopic and statistical techniques for information recovery in metabonomics and metabolomics[J]. Annu Rev Anal Chem (Palo Alto Calif),2008,1:45-69. doi: 10.1146/annurev.anchem.1.031207.113026 [29] BOROUGHS L K, DEBERARDINIS R J. Metabolic pathways promoting cancer cell survival and growth[J]. Nat Cell Biol,2015,17(4):351-359. doi: 10.1038/ncb3124 [30] LIBERTI M V, LOCASALE J W. The Warburg effect: how does it benefit cancer cells?[J]. Trends Biochem Sci, 2016, 41(3): 211-218. [31] KOUNDOUROS N, POULOGIANNIS G. Reprogramming of fatty acid metabolism in cancer[J]. Br J Cancer,2020,122(1):4-22. doi: 10.1038/s41416-019-0650-z [32] TSUN Z Y, POSSEMATO R. Amino acid management in cancer[J]. Semin Cell Dev Biol,2015,43:22-32. doi: 10.1016/j.semcdb.2015.08.002 [33] HASHIM ABDALLA M S, TAYLOR-ROBINSON S D, SHARIF A W, et al. Differences in phosphatidylcholine and bile acids in bile from Egyptian and UK patients with and without cholangiocarcinoma[J]. HPB,2011,13(6):385-390. doi: 10.1111/j.1477-2574.2011.00296.x [34] SHARIF A W, WILLIAMS H R, LAMPEJO T, et al. Metabolic profiling of bile in cholangiocarcinoma using in vitro magnetic resonance spectroscopy[J]. HPB (Oxford),2010,12(6):396-402. [35] MURAKAMI Y, KUBO S, TAMORI A, et al. Comprehensive analysis of transcriptome and metabolome analysis in Intrahepatic Cholangiocarcinoma and Hepatocellular Carcinoma[J]. Sci Rep,2015,5:16294-16305. doi: 10.1038/srep16294 [36] PADTHAISONG S, PHETCHARABURANIN J, KLANRIT P, et al. Integration of global metabolomics and lipidomics approaches reveals the molecular mechanisms and the potential biomarkers for postoperative recurrence in early-stage cholangiocarcinoma[J]. Cancer Metab,2021,9(1):30-44. [37] YI M, LI J J, CHEN S N, et al. Emerging role of lipid metabolism alterations in Cancer stem cells[J]. J Exp Clin Cancer Res,2018,37(1):118-135. [38] LIANG Q, WANG C, LI B B, et al. Lipidomics analysis based on liquid chromatography mass spectrometry for hepatocellular carcinoma and intrahepatic cholangiocarcinoma[J]. RSC Adv,2015,5(78):63711-63718. [39] WANG X X, LI J, ZHANG A H. Urine metabolic phenotypes analysis of extrahepatic cholangiocarcinoma disease using ultra-high performance liquid chromatography-mass spectrometry[J]. RSC Adv,2016,6(67):63049-63057. doi: 10.1039/C6RA09430A [40] HAZNADAR M, DIEHL C M, PARKER A L, et al. Urinary metabolites diagnostic and prognostic of intrahepatic cholangiocarcinoma[J]. Cancer Epidemiol Biomarkers Prev,2019,28(10):1704-1711. [41] BANALES J M, IÑARRAIRAEGUI M, ARBELAIZ A, et al. Serum metabolites as diagnostic biomarkers for cholangiocarcinoma, hepatocellular carcinoma, and primary sclerosing cholangitis[J]. Hepatology,2019,70(2):547-562. doi: 10.1002/hep.30319 [42] MACIAS R I R, MUÑOZ-BELLVÍS L, SÁNCHEZ-MARTÍN A, et al. A novel serum metabolomic profile for the differential diagnosis of distal cholangiocarcinoma and pancreatic ductal adenocarcinoma[J]. Cancers (Basel),2020,12(6):1433-1450. doi: 10.3390/cancers12061433 [43] KOTAWONG K, CHAIJAROENKUL W, ROYTRAKUL S, et al. Screening of molecular targets of action of atractylodin in cholangiocarcinoma by applying proteomic and metabolomic approaches[J]. Metabolites,2019,9(11):260-273. doi: 10.3390/metabo9110260 [44] GIUSTARINI D, GALVAGNI F, TESEI A, et al. Glutathione, glutathione disulfide, and S-glutathionylated proteins in cell cultures[J]. Free Radic Biol Med,2015,89:972-981. doi: 10.1016/j.freeradbiomed.2015.10.410 [45] ZHANG J, HANG C H, JIANG T, et al. Nuclear magnetic resonance-based metabolomic analysis of the anticancer effect of metformin treatment on cholangiocarcinoma cells[J]. Front Oncol,2020,10:570516-570528. doi: 10.3389/fonc.2020.570516 [46] BI L, REN Y D, FENG M X, et al. HDAC11 regulates glycolysis through the LKB1/AMPK signaling pathway to maintain hepatocellular carcinoma stemness[J]. Cancer Res,2021,81(8):2015-2028. doi: 10.1158/0008-5472.CAN-20-3044 [47] STEPHENNE X, FORETZ M, TALEUX N, et al. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status[J]. Diabetologia,2011,54(12):3101-3110. doi: 10.1007/s00125-011-2311-5 [48] CHEN X, LIU H S, SHEN L, et al. Untargeted UPLC-MS-based metabolomics analysis reveals the metabolic profile of intrahepatic cholangiocarcinoma process and the intervention effect of Osthole in mice[J]. Pharmacol Res Mod Chin Med,2022,3:100096-100113. doi: 10.1016/j.prmcm.2022.100096 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 9949
- HTML全文浏览量: 3573
- PDF下载量: 42
- 被引次数: 0
 下载:
下载:
