

 下载:
下载:

-
光动力治疗(PDT)基于光辐照聚集光敏剂的肿瘤组织,由光敏剂诱发光动力反应形成单线态氧(1O2)等活性氧(ROS),通过对肿瘤细胞和肿瘤血管的直接杀伤及激活机体系统免疫反应等多种机制发挥抗肿瘤作用[1-3]。二氢卟吩及菌绿素类光敏剂是PDT新药研究的热点[4-8]。其中,已获批上市的代表药物有他拉泊芬(talaporfin)和帕利泊芬(padeliporfin)等[9, 10]。
光敏剂作为结构非特异性药物,存在缺乏肿瘤靶向性摄入和明确的作用药靶等缺陷。此外,PDT受制于局部治疗,对浸润较深的肿瘤组织,及已发生转移的肿瘤疗效有限。目前,PDT和化疗联用是克服上述缺陷,提高PDT疗效最为普遍和有效的策略之一。研究表明,抗代谢化疗药物氟尿嘧啶(5-Fu)与PDT联用具有协同抗肿瘤作用[11-13]。据此,我们设想利用在肿瘤微环境下能响应性断裂的连接基团(linker)将光敏剂与化疗药物偶联,希望实现二者在肿瘤组织的靶向释放,从而发挥其PDT和化疗协同抗肿瘤作用。酰腙键是酸敏感化学键,常被用来连接载体,以药物制备智能药物载体。这种药物载体到达肿瘤细胞的内涵体或溶酶体中时,会发生酸性水解将药物有效释放出来。因此,本文针对肿瘤微环境呈弱酸性的特点,采用药物化学最经典的前药设计策略,以脱镁叶绿素a(Phephorbide a)粗提物经酸碱降解制得的二氢卟吩e6(3)[14]为先导光敏剂,通过其152-羧基与抗肿瘤药物5-Fu以酸敏感酰腙键连接,设计合成pH响应型光化疗协同抗肿瘤光敏剂二氢卟吩e6-偕氟尿嘧啶(1),并考察其体外PDT抗肿瘤活性和pH响应性5-Fu释放,及其对黑色素瘤B16-F10和肝癌HepG2细胞的光动力抗癌活性及其作用机制,以期获得高效、低毒的PDT治癌药物候选药物,合成路线见图1。
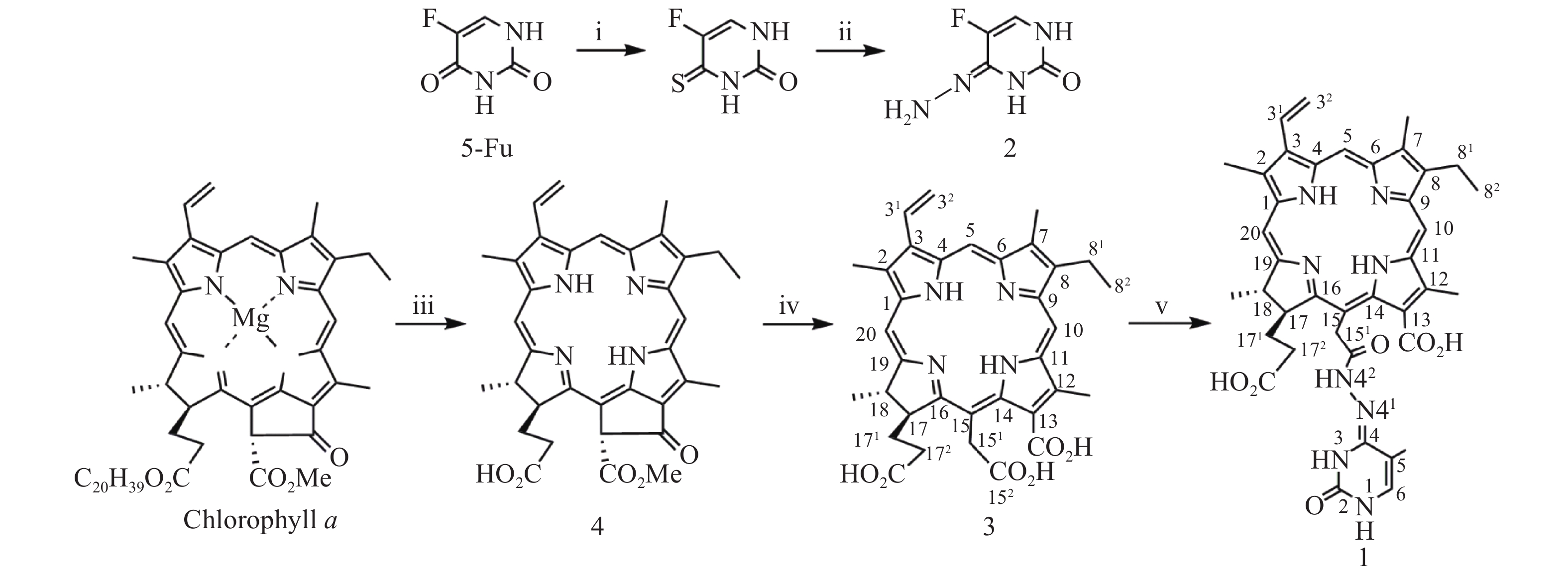
图 1 二氢卟吩e6 -偕氟尿嘧啶光敏剂(1)的合成路线
-
用Bruker MSL-600型核磁共振仪测定1H NMR,CD3OD为溶剂;用API-3000 LC-MS型电喷雾质谱仪测定质谱(ESI-MS);用岛津UV-160型紫外分光光度计测定UV吸收谱;用日立F-7000荧光分光光度计测定荧光发射谱;用Shimazu LC-20AD HPLC仪测定化合物1的相对纯度及其5-Fu的体外释放。色谱柱型号为Waters Xterra C18柱,流动相:乙腈-0.3%乙酸水溶液(80 : 20);流速:1.0 ml/min;检测波长:400 nm(化合物1的相对纯度)或254 nm(5-Fu释放);柱温:30 ℃;进样量:20 μl。柱色谱分离用TELEDYNE ISCO的快速制备色谱Combi Flash@Rf+仪,硅胶H作为固定相。PDT抗癌活性测试使用BWT半导体激光仪(北京凯普林,波长为660 nm);用流式细胞仪(BD Accuri C6,美国)(激发波长:488 nm,发射波长:525 nm)检测受试肿瘤细胞样品的ROS水平、细胞凋亡率和细胞周期阻滞。
二氢卟吩e6(3)按照文献[14]的方法制备;其它实验用材料和化学试剂均为市售商品。
-
取氟尿嘧啶(0.2 g,1.563 mmol)溶于无水吡啶(10 ml),加入五硫化二磷(0.298 g,1.563 mmol),加热回流12 h。反应完毕,减压回收溶剂,残物加乙酸乙酯溶解(100 ml),用0.1 mol/L HCl洗涤(50 ml×2),无水Na2SO4干燥,减压除去溶剂得4-硫代-5-氟尿嘧啶粗品。上述4-硫代-5-氟尿嘧啶粗品加甲醇(10 ml)溶解,于0 ℃下滴加N2H4·H2O(0.316 g,6.252 mmol),室温继续搅拌2 h。反应完毕,减压抽滤,P2O5真空干燥得固体化合物5-氟尿嘧啶-4-腙(2)中间体,直接用于下步反应。取二氢卟吩e6(0.1 g,0.168 mmol)溶于无水DMF(10 ml),加1-乙基-(3-二甲氨基丙基)碳二亚胺盐酸盐(EDC·HCl)(0.035 g,0.183 mmol),室温搅拌反应6 h后再加入中间体2(0.031 g,0.218 mmol),继续搅拌36 h。反应完毕,反应液加入10倍体积量乙酸乙酯,饱和NaCl水溶液洗涤(50 ml×3),无水Na2SO4干燥,减压回收溶剂所得固体经快速制备色谱梯度洗脱分离纯化(流动相为二氯甲烷/甲醇/甲酸=15∶1∶0.1~8∶1∶0.1)得黑色固体1纯品0.048 g,产率39.6%。UV-vis λmax (MeOH, nm) (ε, M−1cm−1):660 (3.15×104), 510 (0.82×104), 402 (8.13×104)。1H-NMR (600 MHz, CD3OD, δ, ppm): 9.79 (s, 1H, 10-CH), 9.73 (s, 1H, 5-CH), 9.07 (s, 1H, 20-CH), 8.19 (dd, J = 18.0, 12.0 Hz, 1H, 31-CH), 7.29 (s, 1H, 5-Fu的6-CH), 6.38 (d, J = 18.0 Hz, 1H, 32-CHB), 6.15 (d, J = 12.0 Hz, 1H, 32-CHA), 5.35 (s, 2H, 151-CH2), 4.65 (m, 2H, 17-CH和18-CH), 3.84 (q, J = 7.5 Hz, 2H, 81-CH2), 3.63 (s, 3H, 12-CH3), 3.53 (s, 3H, 2-CH3), 3.30 (s, 3H, 7-CH3), 2.3~2.0 (m,4H , 171-CH2 和172-CH2), 1.76 (m, 6H¸ 18-CH3和82-CH3)。MS (ESI+) m/z: 723.63 (M+H)+ (100%)。元素分析(C38H39N8O6F,%)计算值:C 63.16, H 5.40, N 15.48;实测值:C 63.34, H 5.38, N 15.43。HPLC测定纯度:95.2%。
-
分别测定目标化合物1及其先导化合物二氢卟吩e6(3)的甲醇溶液(10 μmol/L)在300~800 nm处的紫外吸收谱和激发波长为400 nm的荧光发射光谱,结果见图2。
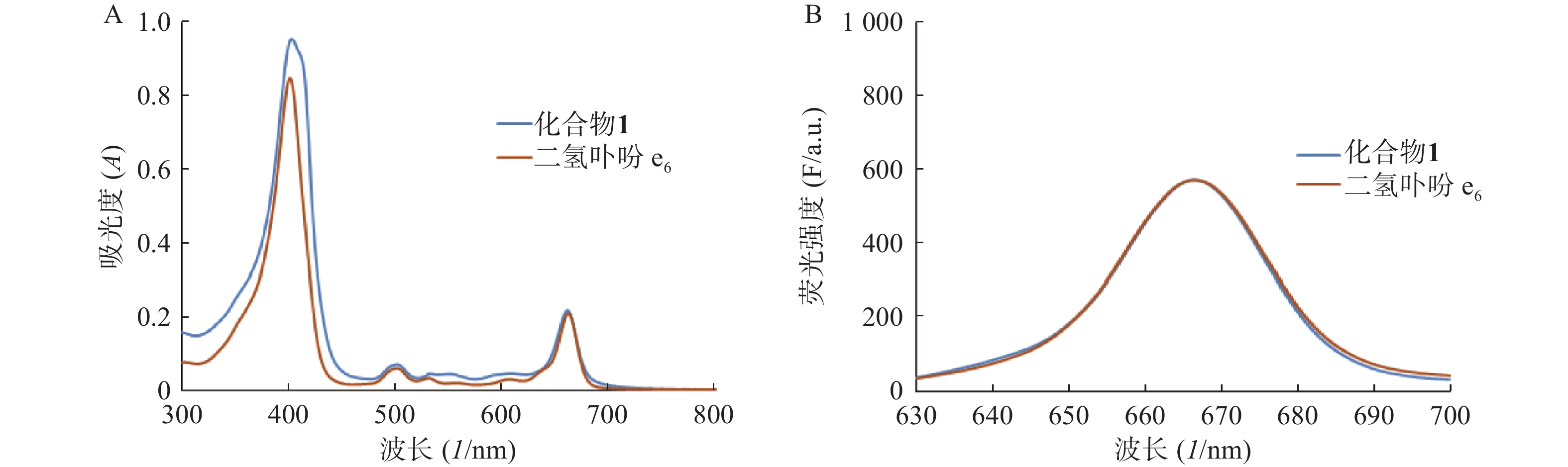
图 2 化合物1甲醇液(10 μmol/L)的紫外吸收谱和荧光发射谱(λEx=400 nm)
-
分别配制浓度为50 μmol/L的化合物1的HOAc-NaOAc缓冲液(pH 5.0)和PBS溶液(10 ml),并于0.5、1.0、3.0、6.0、12、24 h时分别取样(500 μl)。其中,HOAc-NaOAc缓冲液(pH 5.0)组取样液用0.1 mol/L氢氧化钠水溶液迅速调节pH值至7.4。每份取样液加PBS稀释至原溶液1/3浓度,微孔滤膜(孔径0.22 μm)过滤,HPLC进样检测;实验重复3次。根据5-Fu的HPLC峰面积-浓度标准曲线分析计算,绘制目标化合物1于弱酸(pH 5.0)中的5-Fu体外释放量-时间曲线,结果见图3。
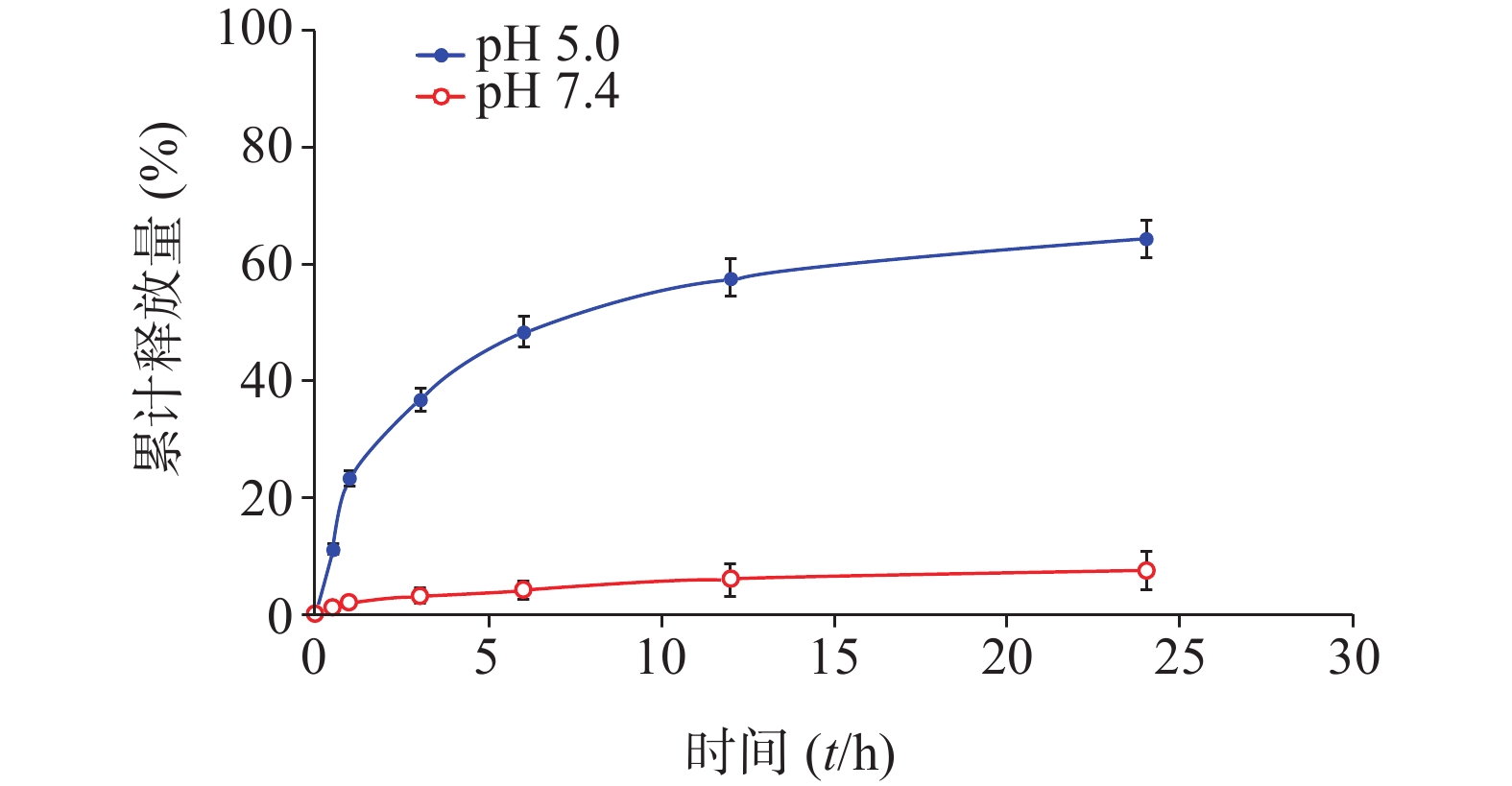
图 3 化合物1的体外pH响应性5-Fu累积释放量-时间曲线(n=3)
-
参照文献[6-8]的方法,每孔5×103个B16-F10细胞或HepG2细胞悬液(100 μl)接种于96孔板上,加入等体积上述细胞培养液孵育24 h;更换含不同浓度待测物的培养液(DMSO浓度小于1%,100 μl),继续避光孵育48 h;再更换含10%(V/V)CCK-8(Beyotime,中国)的RPMI 1640基础培养基(100 μl),继续培养1.5 h,然后用Varioskan Flash全波长酶标仪(Thermo)于波长450 nm处测定每孔的吸光度值,计算各浓度对应的细胞存活率,并拟合得到待测物的肿瘤细胞半数抑制浓度即IC50值。
-
每孔5×103个B16-F10细胞或HepG2细胞悬液(100 μl)接种于96孔板上,加入等体积细胞培养液孵育24 h;更换含不同浓度待测物的细胞培养液(DMSO浓度小于1%,100 μl),继续避光孵育24 h;再更换新鲜培养液(100 μl),以波长为660 nm的激光辐照受试细胞样品(光照剂量为10 J/cm2),继续孵育24 h。最后按“2.3.2”项下CCK-8法测定各待测物的肿瘤细胞IC50值。
-
以临床光敏药物他拉泊芬为阳性对照,化合物1及其先导化合物3对肿瘤细胞株的体外PDT抗癌活性结果见表1。
表 1 目标化合物1的体外光动力抗癌活性(IC50,μmol/L)
化合物 B16-F10细胞 暗毒/光毒比 HepG2细胞 暗毒/光毒比 暗毒性 光毒性 暗毒性 光毒性 化合物 1 46.84±8.46*, ΔΔΔ 0.73±0.16**, ΔΔΔ 64.2 50.80±6.45**, #, ΔΔΔ 0.90±0.22**, ΔΔΔ 56.4 二氢卟吩e6 69.72±4.69 3.36±0.59 20.8 70.38±10.9 2.75±0.41 25.6 他拉泊芬 254.8±18.8 11.31±3.88 22.5 176.4±28.4 15.47±5.07 11.4 5-Fu 35.80±6.68 NTa − 39.16±2.7 NTa − NTa:未测定;*P < 0.05,**P < 0.01,与二氢卟吩 e6组比较;#P < 0.05,与5-Fu组比较;ΔΔΔP < 0.001,与他拉泊芬组比较。 -
操作步骤如下:a. 每孔3 × 105个B16-F10细胞悬液(2 ml)接种6孔板上,按“2.3.1”项条件避光孵育24 h;b. 分别更换含一定浓度化合物1或他拉泊芬的新鲜培养液(DMSO浓度小于1%,2 ml),继续避光孵育24 h;c. 加入10 mmol/L DCFH-DAROS荧光检测探针(Beyotime,1.5 μl),吹打混匀,继续避光孵育20 min;d. PBS洗涤3次,再加新鲜培养液(2 ml),以660 nm波长的激光辐照(光剂量10 J/cm2)细胞样品,继续避光孵育20 min;e. 收集每孔细胞样品,用流式细胞仪检测各孔细胞ROS水平,结果见图4。
图 4 目标化合物1诱导B16-F10细胞产生活性氧的水平
-
按“2.4”项下操作方法,仅从步骤c开始,更换新鲜培养液(2 ml),用660 nm波长的激光辐照(光剂量10 J/cm2)细胞样品,继续避光孵育20 min;d. 以1 500 r/min离心(5 min)细胞样品,PBS洗涤,再以1 000 r/min离心(5 min)后获取细胞样品;e. 按Annexin V-FITC细胞凋亡检测试剂盒(Beyotime)操作流程操作,结果见图5。
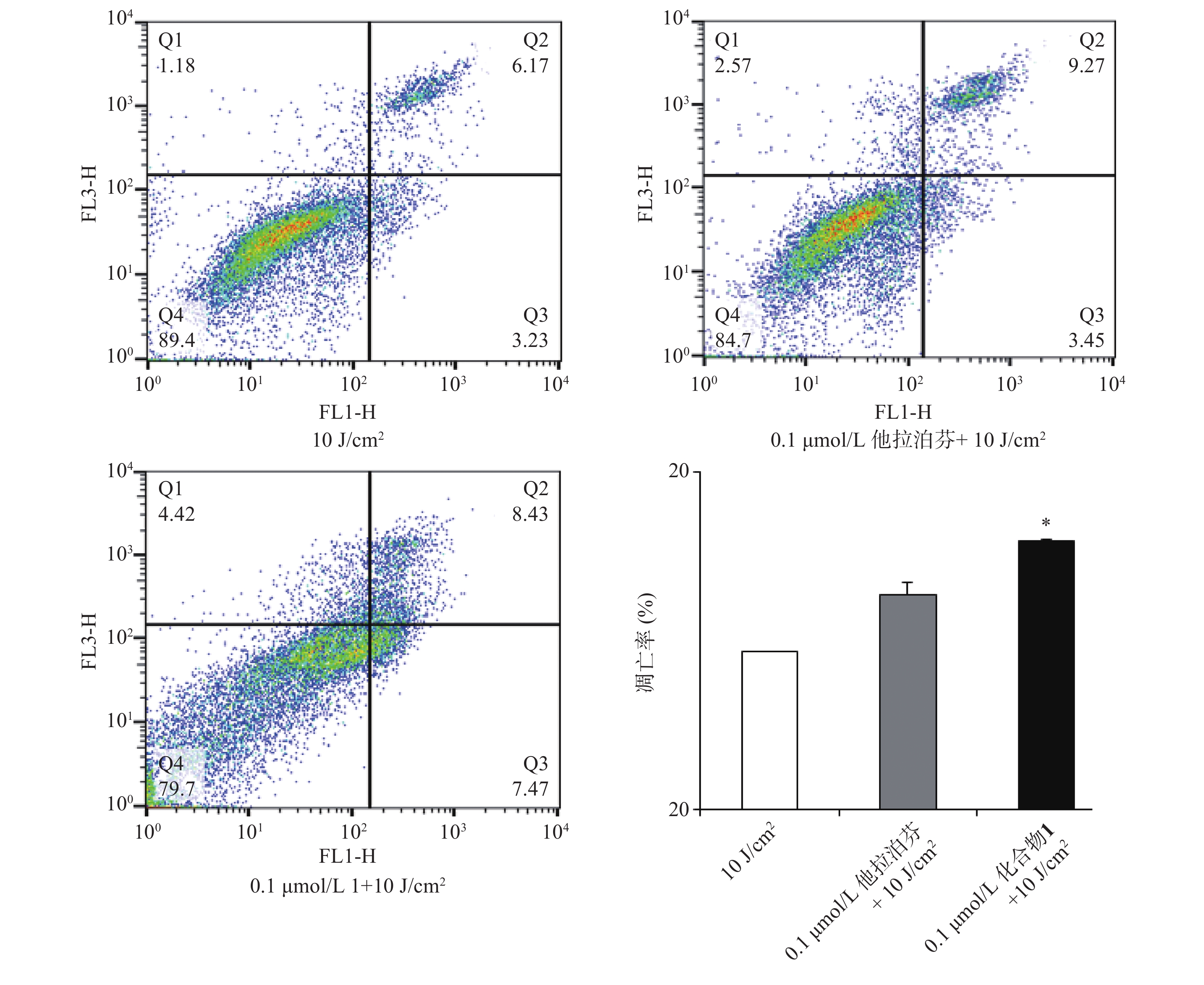
图 5 目标化合物1诱导B16-F10细胞凋亡
-
按“2.5”项下操作方法,仅在e步骤中,换以细胞周期阻滞检测试剂盒(Beyotime)的操作流程,每份细胞样品中分别加入染色缓冲液(300 µl)、RNase A(6 µl)和碘化丙啶染色液(15 µl),轻轻混匀,避光孵育20 min后,用流式细胞仪进行细胞周期阻滞检测,结果见图6。
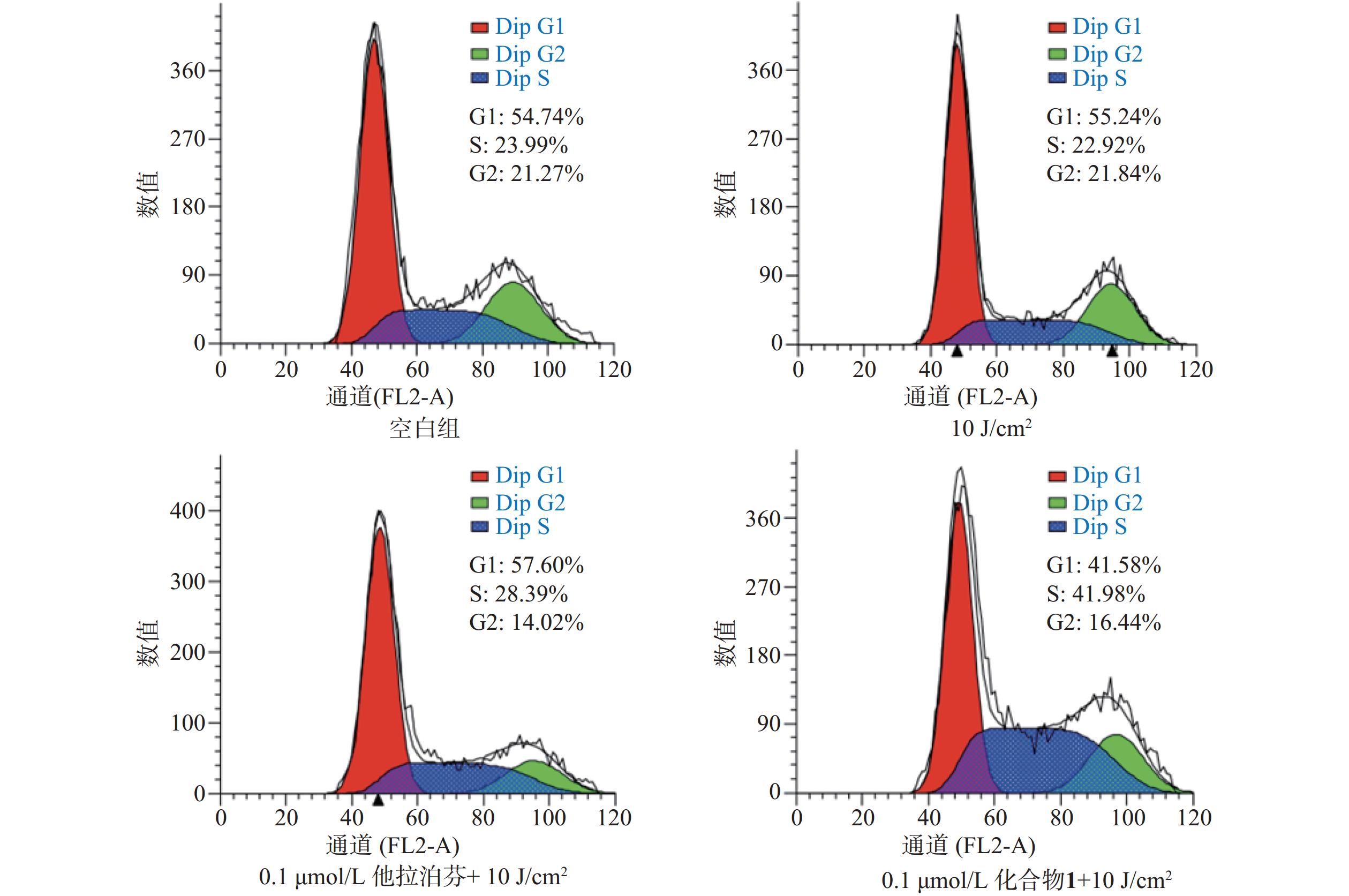
图 6 目标化合物1对B16-F10细胞周期的阻滞作用
-
按文献[14]方法制得的二氢卟吩e6(3)为先导化合物,经1-乙基-(3-二甲氨基丙基)碳二亚胺盐酸盐(EDC·HCl)于无水DMF中催化分子内脱水缩合制得二氢卟吩e6-131,152-酸酐活泼中间体[15],然后直接与中间体2发生酰化反应成功合成得到了光化疗双模抗肿瘤光敏剂二氢卟吩e6-偕氟尿嘧啶(1),反应收率达39.6%,其结构经UV、ESI-MS、1H NMR及元素分析确证。
化合物1在甲醇中最大紫外吸收波长和荧光发射波长(激发波长:400 nm)分别为660 nm和670 nm,与先导物3相一致,表明先导物3以酰腙键偶联5-Fu后,并没有改变其作为光敏剂特有的紫外吸收和荧光发射光谱等光物理特性。此外,化合物1在弱酸(pH 5.0)条件下,能有效释放5-Fu,24 h内累积释放率可达60.3%;但在pH 7.4的条件下较为稳定,24 h内5-Fu累积释放率仅为5%。
体外PDT抗癌活性测试结果显示,化合物1对B16-F10和HepG2细胞株的光毒活性和暗毒/光毒比(治疗指数)均显著优于先导物二氢卟吩e6(3)(P<0.005)和他拉卟吩(P<0.001),其IC50值分别达0.73 μmol/L和0.90 μmol/L。
体外PDT抗癌机制研究提示,化合物1介导的PDT能显著提升B16-F10细胞内ROS水平和诱导B16-F10细胞凋亡,并阻滞肿瘤细胞周期于S期。
总之,二氢卟吩e6-偕氟尿嘧啶(1)具有PDT抗癌活性强、治疗指数(暗毒/光毒比)高且可在肿瘤弱酸环境中有效释放5-Fu等优点,从而实现“单分子”光化疗协同抗肿瘤作用,值得进一步开发研究。
Synthesis and biological activities of chlorin e6-based conjugate of fluorouracil as dual-mode antitumor photosensitizer
-
摘要:
目的 文献报道氟尿嘧啶(5-Fu)与光敏剂联用具有协同抗肿瘤作用,笔者设计合成二氢卟吩e6(化合物 3 )与5-Fu经酰腙键偶联的pH响应性、光化疗双模抗肿瘤光敏剂(化合物 1 ),研究其初步体外光动力抗癌活性及作用机制。 方法 首先,5-Fu与五硫化二磷于吡啶中回流反应形成4-硫代-5-氟尿嘧啶,再和水合肼于甲醇中反应制得5-氟尿嘧啶-4-腙(化合物 2 );然后,将脱镁叶绿素a(化合物 4 )酸碱降解产物 3 经EDC·HCl催化缩合形成二氢卟吩e6-131,152-酸酐中间体后,直接与 2 发生选择性酰化反应,制得目标化合物 1 ,并考察其体外pH响应性5-Fu释放及对黑素瘤B16-F10和肝癌HepG2细胞的光动力抗癌活性和作用机制。 结果 化合物 1 在微酸(pH 5.0)环境中能有效释放5-Fu,24 h累积释放率可达60.3%;其在光照下对黑素瘤B16-F10和肝癌HepG2细胞的半数抑制浓度(IC50)分别为0.73 μmol/L和0.90 μmol/L,均显著优于先导物 3 和上市药物他拉泊芬(talaporfin),且能显著提升肿瘤细胞内活性氧(ROS)水平和诱导肿瘤细胞凋亡,并阻滞肿瘤细胞周期于S期。其结构经紫外、电喷雾质谱、氢谱和元素分析确证。 结论 新型双模抗肿瘤光敏剂化合物 1 具有光动力抗癌活性强、治疗指数(暗毒/光毒比)高,且可在微酸(pH 5.0)环境响应性释放5-Fu等优点,从而实现“单分子”光化疗双重抗肿瘤作用,值得进一步开发研究。 Abstract:Objective To design and synthesize the conjugate (compound 1 ) of chlorin e6 (compound 3 ) with fluorouracil (5-Fu) as novel pH-responsive dual-mode antitumor photosensitizer by acyl hydrazone bond coupling, based on literature reports that combination of 5-Fu and photosensitizer possess synergistic anti-tumor effect, and investigate its photodynamic antitumor activity and mechanism. Methods Lead compound 3 was obtained by alkali degradation with 25% KOH-CH3OH on pheophorbide a (compound 4 ) which was prepared through acid hydrolysis of chlorophyll a in crude chlorophyll extracts from silkworm excrement. Reflux reaction of 5-Fu with P2S5 in pyridine formed crude 4-thio-5-fluorouracil which was followed to react with hydrazine hydrate (N2H4·H2O) in CH3OH to give 5-fluorouracil-4-hydrazone (compound 2 ). Then, treatment of compound 3 i.e. acid alkali degradation product of chlorophyll a in silkworm excrement with EDC·HCl generated its 171- and 152 cyclic anhydride which was followed to directly react with intermediate compound 2 to successfully get title compound 1 . In addition, its pH-responsive 5-Fu release and photodynamic antitumor activity and their mechanisms in vitro were investigated. Results Compound 1 could responsively release 5-Fu at pH 5.0, with a cumulative release rate of 60.3% within 24 h. It exhibited much higher phototoxicity against melanoma B16-F10 and liver cancer HepG2 cells than talaporfin and its precursor compound 3 , with IC50 value being 0.73 μmol/L for B16-F10 cells and 0.90 μmol/L for HepG2 cells, respectively. Upon light irradiation, it also could significantly induce cell apoptosis and intracellular ROS level and block cell cycle in S phase. Its structure was confirmed by UV, 1H-NMR, ESI-MS and elemental analysis data. Conclusion The conjugate compound 1 of compound 3 and 5-Fu has the advantages of strong PDT anticancer activity, high therapeutic index (i.e. dark toxicity/phototoxicity ratio) and responsively release 5-Fu at pH 5.0 etc. which shows “unimolecular” dual antitumor effects of PDT and chemotherapy and is worthy of further research and development. -
Key words:
- synthesis /
- photodynamic therapy /
- photosensitizer /
- chlorin e6 /
- fluorouracil (5-Fu) /
- antitumor
-
创伤大出血是人类创伤死亡的第二大死因,占创伤死亡总数的15%[1]。在院前止血阶段,传统止血材料在一定程度上存在止血效果不佳、储存时间短、应用不方便等缺点,使得控制创伤大出血仍是一大亟待解决的难题。进入到院内手术阶段,简单的纱布止血仍是一种常规选择,这为开发新型止血材料提供了广阔的空间。纳米技术可以在纳米尺度上改造并利用微观结构,赋予了纳米材料改良的扩散性和溶解性、易于穿透生理屏障、比表面积大、药物的缓控和靶向释放等独特优势[2]。近年来,基于脂质体、纳米粒、自组装纳米肽等纳米止血材料的研究日益深入,为现代化新型止血材料的发展奠定了良好基础。本文旨在综述脂质体、纳米粒、自组装纳米肽、纳米纤维等多种纳米止血材料的前沿设计和应用进展,为下一步研究应用提供参考。
1. 脂质体
脂质体是一种研究广泛的纳米递送系统。它通常由磷脂和胆固醇制备而成,形成磷脂分子亲水头部插入亲水介质,疏水尾部伸向疏水介质的球形结构,直径大小一般在20 nm到10 µm[3]。脂质体的性质随脂质种类、表面电荷、粒径大小和制备方法的不同而有很大差异[4]。在止血方面,脂质体可以包裹止血生物大分子,提高其稳定性和生物相容性,降低生物大分子的副作用;脂质体也可偶联止血多肽链,在增强多肽链稳定性的同时发挥其止血效果。根据脂质体发挥功能的不同,可以将其大致分为止血生物大分子内载脂质体和止血多肽链修饰脂质体[5]。Chan等将凝血酶包裹到纳米脂质体中,并通过体外监测血小板活化、血块收缩等实验来评估其凝血功能。研究结果表明,装载凝血酶的脂质体能被血小板内吞利用,使这种血小板对激动剂更加敏感[6]。Nishikawa等开发了一种纤维蛋白原γ链修饰并内载二磷酸腺苷(adenosine diphosphate,ADP)的脂质体。该脂质体平均直径为210 nm,通过糖蛋白GⅡb/Ⅲa与活化的血小板相互作用以及ADP对血小板的增强聚集来实现静脉治疗患急性血小板减少症兔肝出血模型的有效止血,并且在动物肺部、肾脏和肝脏未检测到血栓形成[7]。Hickman等评估了血管性血友病因子结合肽、胶原结合肽以及纤维蛋白原模拟肽修饰的脂质体对治疗猪股动脉出血的止血效能。实验结果表明,该脂质体处理的猪在前30 min的失血率显著低于对照组,并最终实现了完全止血。处理组动物的股动脉血块富含掺杂脂质体的血小板,而在其他器官组织样本中没有发现血栓形成[8]。
2. 纳米粒
纳米粒指由大分子物质组成的固体胶体颗粒,其粒径大小通常为10~1000 nm[9]。天然与合成聚合物的纳米粒载药稳定性良好,且易于表面修饰,并可以通过调节聚合物的特性和表面修饰来实现药物的可控释放和靶向定位[10]。带电荷的纳米颗粒能与带相反电荷的血细胞或者纤维蛋白原产生静电作用,中和表面电荷后诱导其聚集,促进血液的凝固[11]。Biranje等采用离子凝胶法制备壳聚糖纳米颗粒,通过冷冻干燥将其组装成多孔壳聚糖敷料。该壳聚糖敷料平均孔径为4.074 nm,比表面积为61.83 m2/g,具有孔隙率高、溶胀性好、生物降解性好、生物相容性好等特点,有利于促进止血和创面愈合[12]。Meddahipelle等研究证明了二氧化硅与氧化铁纳米溶液经过纳米桥联过程可以在1 min内实现大鼠皮肤和肝脏伤口的止血和组织修复[13]。Kudela等研究制备了一种多磷酸盐功能化的二氧化硅纳米颗粒,并证明了将多磷酸盐附着于二氧化硅纳米颗粒可以产生显著增强止血的协同效应,缩短凝血时间。这种多磷酸盐功能化的二氧化硅纳米颗粒可以增强损伤部位的靶向性,最大限度地减少血栓形成并发症的风险[14]。Sundaram等合成了一种平均直径约14 nm的生物玻璃纳米颗粒,并将其掺入壳聚糖水凝胶中,制备了复合水凝胶。该水凝胶具有良好的剪切稀释性和可注入性,在体内、外凝血实验中表现出快速有效的凝血作用。这种生物玻璃纳米颗粒细胞毒性低,血液相容性好,是一种有潜力的创伤止血材料[15]。Gkikas等研究制备了纤维蛋白功能序列甘氨酰-精氨酰-甘氨酰-天门冬氨酰-丝氨酸(Gly-Arg-Gly-Asp-Ser,GRGDS)功能化的聚乳酸羟基乙酸/聚乙二醇(PLGA-PEG)纳米颗粒。研究结果表明,这种生物相容性良好的纳米颗粒静脉注射后会积累在啮齿动物受损肝脏的血凝块中,从而减少失血并显著提高存活率。PLGA-PEG-GRGDS纳米粒通过生物素、1,1'-双十八烷基-3,3,3',3'-四甲基吲哚二碳菁高氯酸盐(DiD)细胞膜荧光染料和金标记后,可以借助共聚焦显微镜、免疫组织化学和CT成像来辅助诊断内出血[16]。
3. 自组装纳米肽
自组装纳米肽(self-assembled nanopeptides,SAP)是指将相对简单的肽链通过非共价自组装而形成的有序纳米结构肽。它不仅能用于药物递送,而且能在体内任何潮湿的离子环境中形成一种纳米纤维屏障,并能浓缩血液有形成分来控制出血。自组装纳米肽生物相容性良好,能在生物体内分解成天然氨基酸,可以被周围组织用于修复。Ellis-Behnke等用自组装纳米短肽RADA16-I制备了一系列不同质量浓度的溶液。在脑、股动脉和肝切口的小鼠模型中,局部使用不同浓度的溶液治疗均能显著缩短止血时间。电子显微镜显示,该溶液会自组装成屏障,阻止血液流动并促进相邻细胞的移动来修复受损部位。这种自组装纳米短肽无毒、无免疫原性,并且其降解产物是氨基酸,可用于组织修复,是一种良好的止血材料[17]。Cheng等制备了纤维蛋白功能序列(GRGDS)与层黏连蛋白功能序列酪氨酰-异亮氨酰-甘氨酰-丝氨酰-精氨酸(Tyr-Ile-Gly-Ser-Arg,YIGSR)的自组装纳米肽,并研究证明了该纳米肽具有良好的生物相容性与局部止血效果,并可显著促进肝组织再生[18]。Morgan等将3种组织因子特异性结合多肽序列(EGRNCETHKDDQL,RLMTQDCLQQRSK,RTLAFVRFK序列)共价结合到两亲性肽链骨架,自组装成3种纳米肽纤维。研究发现,只有RTLAFVRFK序列所结合的纳米肽纤维才可以显著减少失血量,且增加纤维的密度可以增强止血效果。生物相容性实验表明,该纳米肽纤维不会诱导红细胞溶血,不会在肝损伤部位诱导炎症,并在血浆中30 min后仍有70%材料保持结构的完整性[19]。
4. 纳米纤维
纳米纤维通常指直径为1~100 nm,并且具有一定长度的线状纳米材料。它可以通过静电纺丝技术从各种天然与合成聚合物中提取制备而成。纳米纤维具有比表面积大、可调节的孔隙率和易表面功能化等优点,使其在抗菌止血敷料、给药系统以及组织工程等生物医学领域被广泛应用。特殊材料的电纺纳米纤维可形成纳米纤维垫,具有比表面积大和孔隙率高的优点,并能与止血药物共混应用,最终达到快速止血的效果[20]。Yin等研究开发了基于季铵化N-卤胺壳聚糖和聚乙烯醇的新型抗菌止血纳米纤维膜。该电纺膜具有均匀的纳米纤维结构,孔隙率高,与细胞外基质相似,具有优异的吸水性能和良好的力学性能。细胞相容性实验结果表明,人成纤维细胞可以在这种膜上黏附并增殖,从而证明了其良好的生物相容性。在全血凝固实验中,该膜表现出良好的凝血活性,不仅具有显著的血浆吸附性,而且能诱导血小板黏附和活化[21]。Liu等将氨基化纳米银和明胶引入羧化纤维素纳米纤维中,成功制备了一种纳米复合水凝胶。该复合水凝胶具有较强的机械性能、抗菌性能和良好的止血性能,在体内外创面愈合模型评价中显示出良好的生物相容性和创面愈合效果[22]。Dong等设计了一种以氰基丙烯酸酯为原料,用于内脏止血的气体辅助原位电纺丝装置。该装置可以提高氰基丙烯酸酯聚合物的沉积精度,避免组织黏连;辅助气流可以将聚合纤维吹到组织表面,可在几秒钟内完成肝脏止血[23]。Chen等以聚己内酯为原料,制备出一种可注射的聚己内酯花生状纳米纤维颗粒。该纳米纤维颗粒可通过套管或注射器输送到受伤部位,与血液接触后几秒内重新膨胀到原来的形状,能有效控制出血。此外,涂覆明胶层的花生状纳米纤维颗粒显示出比商用纱布和Gelfoam®更好的血液凝血效果[24]。Sasmal等以聚乙烯醇和壳聚糖为原料,通过静电纺丝技术制备了聚乙烯醇/壳聚糖复合纳米纤维膜,并将止血药氨甲环酸负载其上。这种纳米纤维膜显示出良好的血液相容性与抗生物膜形成性能,并且纤维膜中的壳聚糖成分显著增强了氨甲环酸的止血效果[25]。快速止血剂mRDH由乙酰氨基葡萄糖纳米纤维材料组成,具有血管收缩,血小板活化,红细胞聚集等止血机制。mRDH创伤绷带可有效控制严重内脏损伤和肝破裂的出血,已被FDA批准用于军事和民用环境中快速控制肢体创伤的出血[26]。
5. 结论与展望
纳米材料因其独特的优势,在止血方面有着广泛的研究和应用价值。脂质体、纳米粒、自组装纳米肽等可以通过外部修饰和内部负载止血材料实现良好的止血效果。然而,由于纳米材料出现时间短,评价宏观物质的体系尚难以全方面衡量纳米止血材料的潜在安全性,这使得纳米材料在止血方面的应用受到了很大的限制。虽然纳米止血材料存在诸多未解难题,但不妨碍研究工作者在此领域继续研究拓展。研究人员首先需要从止血机制入手,着力探究机体内细胞分子层面的止血机制,为开发新型纳米止血材料提供机制保证。此外,材料工程是纳米止血材料的基础,开发新型天然与合成的止血化合物并深入研究其止血机制是开发新型纳米止血材料的关键一环。最后,全面系统地评价止血材料的安全性(生物相容性、细胞遗传毒性等)是运用纳米止血材料的底线。研究工作者应不断致力于开发纳米止血材料安全性评价体系,深入研究机体止血机制,增强止血材料与操作者的交互性,最终研发出一种安全、便捷、高效的新型纳米止血材料。
-
图 1 二氢卟吩e6 -偕氟尿嘧啶光敏剂(1)的合成路线
试剂和反应条件:(i)五硫化二磷,吡啶,回流12 h;(ii)水合肼,甲醇,室温2 h;(iii)浓盐酸,乙醚,4 ℃ 30 min;(iv)25%氢氧化钾甲醇液,回流30 min;(v)a. EDC·HCl,N,N-二甲基甲酰胺,室温8 h;b.二异丙基乙胺,2, N,N-二甲基甲酰胺,室温12 h。
表 1 目标化合物1的体外光动力抗癌活性(IC50,μmol/L)
化合物 B16-F10细胞 暗毒/光毒比 HepG2细胞 暗毒/光毒比 暗毒性 光毒性 暗毒性 光毒性 化合物 1 46.84±8.46*, ΔΔΔ 0.73±0.16**, ΔΔΔ 64.2 50.80±6.45**, #, ΔΔΔ 0.90±0.22**, ΔΔΔ 56.4 二氢卟吩e6 69.72±4.69 3.36±0.59 20.8 70.38±10.9 2.75±0.41 25.6 他拉泊芬 254.8±18.8 11.31±3.88 22.5 176.4±28.4 15.47±5.07 11.4 5-Fu 35.80±6.68 NTa − 39.16±2.7 NTa − NTa:未测定;*P < 0.05,**P < 0.01,与二氢卟吩 e6组比较;#P < 0.05,与5-Fu组比较;ΔΔΔP < 0.001,与他拉泊芬组比较。  下载: 导出CSV
下载: 导出CSV
-
[1] AGOSTINIS P, BERG K, CENGEL K A, et al. Photodynamic therapy of cancer: an update[J]. CA Cancer J Clin, 2011, 61(4):250-281. doi: 10.3322/caac.20114 [2] ABRAHAMSE H, HAMBLIN M R. New photosensitizers for photodynamic therapy[J]. Biochem J, 2016, 473(4):347-364. doi: 10.1042/BJ20150942 [3] DOUGHERTY T J. An update on photodynamic therapy appli-cations[J]. J Clin Laser Med Surg, 2002, 20(1):3-7. doi: 10.1089/104454702753474931 [4] DROGAT N, GADY C, GRANET R, et al. Design and synthe-sis of water-soluble polyaminated chlorins and bacteriochlorins - with near-infrared absorption[J]. Dyes Pigments, 2013, 98(3):609-614. doi: 10.1016/j.dyepig.2013.03.018 [5] 刘明辉, 刘俊宏, 韩贵焱, 等. 二氢卟吩p6-13, 15-环酰亚胺类光敏剂的设计合成[J]. 药学实践杂志, 2017, 35(1):26-30,35. [6] MENG Z, YU B, HAN G Y, et al. Chlorin p6-based water-soluble amino acid derivatives as potent photosensitizers for photodynamic therapy[J]. J Med Chem, 2016, 59(10):4999-5010. doi: 10.1021/acs.jmedchem.6b00352 [7] ZHANG X J, MENG Z, MA Z Q, et al. Design and synthesis of novel water-soluble amino acid derivatives of chlorin p6 ethers as photosensitizer[J]. Chinese Chem Lett, 2019, 30(1):247-249. doi: 10.1016/j.cclet.2018.04.029 [8] 马福家, 孟志, 张星杰, 等. 二氢卟吩p6醚类光敏剂的合成及光动力抗癌活性研究[J]. 药学实践杂志, 2020, 38(1):52-56. [9] 张丹萍, 陈志龙, 杨晓霞, 等. 光动力药物的研究与开发[J]. 药学进展, 2007, 31(12):529-535. doi: 10.3969/j.issn.1001-5094.2007.12.001 [10] 闵祥燕, 曹宁, 严懿嘉, 等. 光动力新药帕利泊芬研究进展[J]. 药学进展, 2019, 43(3):231-237. [11] TAHMASEBI H, KHOSHGARD K, SAZGARNIA A, et al. Enhancing the efficiency of 5-aminolevulinic acid-mediated photodynamic therapy using 5-fluorouracil on human melanoma cells[J]. Photodiagn Photodyn, 2016, 13:297-302. doi: 10.1016/j.pdpdt.2015.08.011 [12] ZHAO H Y, YIN R, WANG Y, et al. Modulating mitochon-drial morphology enhances antitumor effect of 5-ALA-mediated photodynamic therapy both in vitro and in vivo[J]. J Photoch Photobio B, 2017, 176:81-91. doi: 10.1016/j.jphotobiol.2017.09.017 [13] ZHANG L L, JI Z J, ZHANG J, et al. Photodynamic therapy enhances skin cancer chemotherapy effects through autophagy regulation[J]. Photodiagn Photodyn, 2019, 28:159-165. doi: 10.1016/j.pdpdt.2019.08.023 [14] 姚建忠, 沈卫镝, 陈文晖, 等. 二氢卟吩e6的合成及其光敏化力和肿瘤光生物活性[J]. 中国医药工业杂志, 2000, 31(5):215-217. doi: 10.3969/j.issn.1001-8255.2000.05.009 [15] CHEN H, WARUNA JINADASA R G, JIAO L J, et al. Chlo-rin e6 131: 152-anhydride: a key intermediate in conjugation reactions of chlorin e6[J]. Eur J Org Chem, 2015, 2015(17):3661-3665. doi: 10.1002/ejoc.201500478 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3415
- HTML全文浏览量: 1409
- PDF下载量: 72
- 被引次数: 0
