

 下载:
下载:
 下载:
下载:

-
对萼猕猴桃Actinidia valvata Dunn是猕猴桃科猕猴桃属中型落叶藤本植物,分布于安徽、浙江、江西等地[1]。其根和根茎作猫人参入药,具有清热解毒、消肿的功效[2],临床常用于治疗肺癌、肝癌及消化道肿瘤[3]。随着猫人参药材的需求量不断增加,在生产过程中,有大量的枝叶未得到有效利用,造成了自然资源的浪费。从中药资源开发利用的角度出发,课题组前期首次对非传统药用部位对萼猕猴桃叶进行了大量研究,发现对萼猕猴桃叶中含有较多的黄酮类成分,具有较强的抗心肌缺血作用和抗氧化活性[4-7]。其中对萼猕猴桃苷E是课题组首次从对萼猕猴桃叶中发现的新黄酮苷类化合物,在叶中含量高,药理活性显著,具有很好的开发前景。作为一个新化合物,如何得到高纯度的对萼猕猴桃苷E并对其进行开发利用,成为一项亟待解决的问题。因此,本文以对萼猕猴桃苷E为研究对象,对其提取分离纯化工艺进行优化,以此获得高纯度的对萼猕猴桃苷E,为后续对萼猕猴桃苷E的药代动力学研究及药理活性研究奠定物质基础。
-
Agilent 1200型高效液相色谱仪(美国安捷伦科技公司);2500Y多功能粉碎机(永康市铂欧五金制品有限公司);BT25S十万分之一电子天平(德国塞利多斯公司);SYG-A2-6电热恒温水浴锅(天津市泰斯特仪器有限公司);SHZ-Ⅲ型循环水真空泵(上海亚荣生化仪器厂);RE-52AA型旋转蒸发器(上海亚荣生化仪器厂);DHG-9123A型电热恒温鼓风干燥箱(上海精宏实验设备有限公司);IS-RDV1恒温振荡器(苏州捷美电子有限公司)。
-
对萼猕猴桃苷E(纯度≥98%,自制);无水乙醇、乙酸乙酯、甲醇为分析纯试剂(上海泰坦科技股份有限公司);甲酸(上海麦克林生化科技股份有限公司)、乙腈(德国Merck公司)、甲醇(上海泰坦科技股份有限公司)为色谱纯试剂;娃哈哈纯净水(杭州娃哈哈集团有限公司);D101型、AB-8型、DM130型、HPD100型、NKA-9型大孔吸附树脂(北京瑞达恒辉科技发展有限公司);柱层析硅胶(100~200目、200~300目,青岛海洋化工有限公司);ODS填料(日本YMC公司)。
-
对萼猕猴桃叶样品为2008年5月采自浙江省衢州市常山县,经海军军医大学辛海量教授鉴定为猕猴桃科猕猴桃属植物对萼猕猴桃Actinidia valvata Dunn的叶。样品于60 ℃烘箱烘干,粉碎过三号筛,得对萼猕猴桃叶粉末,置干燥器中密封保存。
-
色谱柱:Agilent TC-C18(4.6 mm×250 mm, 5 µm);流动相:0.1%甲酸水(A)-乙腈(B)(梯度洗脱:0~35 min, 10%~40% B);流速:1 ml/min;柱温:26 ℃;检测波长:265 nm;进样量:10 µl。对照品及样品色谱图见图1。
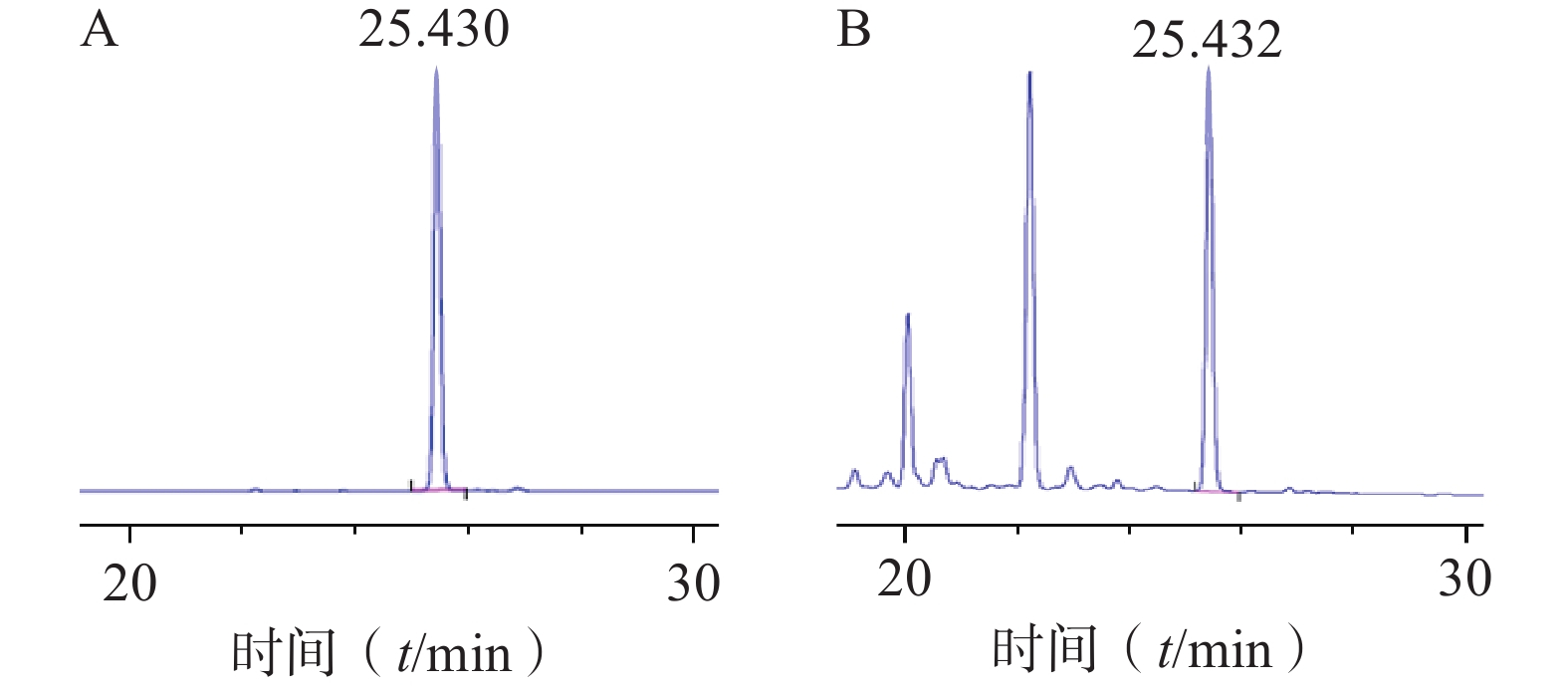
图 1 对萼猕猴桃苷E对照品(A)和对萼猕猴桃叶提取液(B)HPLC图
-
精密称取对萼猕猴桃苷E对照品15.77 mg,置于10 ml容量瓶中,加色谱纯甲醇溶解并定容至刻度,摇匀,即得浓度为1.577 mg/ml的对萼猕猴桃苷E对照品溶液,过0.45 µm微孔滤膜,备用。
-
精密称取对萼猕猴桃叶粉末2 g,置于250 ml圆底烧瓶中,加入55%乙醇50 ml,95 ℃回流提取1 h,过滤后滤液定容至50 ml,摇匀,经0.45 µm微孔滤膜过滤,备用。
-
(1)线性关系考察
精密吸取对萼猕猴桃苷E对照品溶液0.15、0.3、0.6、1.2、2.4、5.0 ml,分别置于5 ml容量瓶中,色谱纯甲醇溶解定容,按“2.1.1”项下色谱条件进样测定。以对萼猕猴桃苷E浓度(mg/ml)为横坐标(c),其峰面积(mAU*s)为纵坐标(A),绘制标准曲线。回归方程为A =
11127 c+31.985,R2=0.9998 ,表明对萼猕猴桃苷E在0.047~1.577 mg/ml范围内与其峰面积线性关系良好。(2)精密度试验
精密吸取同一浓度的对萼猕猴桃苷E对照品溶液连续进样6次,记录其峰面积,相对标准偏差(RSD)为1.29%,表明仪器精密度良好。
(3)稳定性试验
精密称取对萼猕猴桃叶粉末2 g,按“2.1.3”项下方法进行制备,分别于室温0、4、8、12、16、24 h进样测定,记录峰面积,RSD为0.21%,表明供试品溶液在室温条件下24 h内稳定性良好。
(4)重复性试验
称取对萼猕猴桃叶粉末6份,每份2 g,按“2.1.3”项下方法进行制备,进样测定并记录峰面积,RSD为1.07%,表明该方法重复性良好。
(5)加样回收率试验
精密称取已知对萼猕猴桃苷E含量的对萼猕猴桃叶粉末9份,每份1 g,分别加入相当于本底量80%、100%、120%的已知浓度的对萼猕猴桃苷E对照品溶液,按“2.1.3”项下方法进行制备,进样分析并计算加样回收率。对萼猕猴桃苷E的平均加样回收率为101.16%,RSD为1.95%,表明该方法准确可靠。
-
以对萼猕猴桃苷E的含量为指标,单因素考察乙醇浓度、料液比、提取时间、提取温度对对萼猕猴桃苷E含量的影响,并通过四因素三水平L9(34)正交试验优选对萼猕猴桃苷E的最佳提取工艺。
-
(1)乙醇浓度考察
精密称取6份对萼猕猴桃叶粉末,每份5 g,按料液比1∶10分别加入45%、55%、65%、75%、85%、95%乙醇,90 ℃回流提取1 h,滤液定容至50 ml,测定对萼猕猴桃苷E含量。结果表明,对萼猕猴桃苷E的含量随乙醇浓度的增加呈现先升高后降低的趋势,当乙醇浓度为65%时对萼猕猴桃苷E含量最大(图2A)。

图 2 单因素对对萼猕猴桃苷E含量的影响
(2)料液比考察
精密称取6份对萼猕猴桃叶粉末,每份5 g,分别按料液比1∶10、1∶15、1∶20、1∶25、1∶30、1∶35加入65%乙醇,90 ℃回流提取1 h,滤液定容至相应体积。结果表明,料液比过大时,对萼猕猴桃苷E含量下降,其在料液比为1:25时含量最大(图2B)。
(3)提取时间考察
精密称取6份对萼猕猴桃叶粉末,每份5 g,按料液比1:25加入65%乙醇,90 ℃回流提取1次,提取时间分别为0.5、1、1.5、2、2.5、3 h,滤液定容至125 ml。结果表明,对萼猕猴桃苷E含量在提取时间为1 h时最大(图2C)。
(4)提取温度考察
精密称取6份对萼猕猴桃叶粉末,每份5 g,按料液比1:25加入65%乙醇,分别于50、60、70、80、90、100 ℃回流提取1 h,滤液定容至125 ml。结果表明,对萼猕猴桃苷E含量随着提取温度的升高而不断增加,温度升高到90 ℃时,对萼猕猴桃苷E含量骤然提升,当提取温度为100 ℃时其含量最大(图2D)。
-
根据单因素实验考察结果,确定影响对萼猕猴桃苷E提取率的因素为乙醇浓度(A)、料液比(B)、提取时间(C)、提取温度(D)4个因素,每个因素选取3个水平,设计L9(34)正交表进行正交试验,优化对萼猕猴桃苷E的提取工艺。重复试验正交结果及直观分析见表1,方差分析见表2,结果表明,影响对萼猕猴桃苷E含量的主次因素为提取温度>乙醇浓度>料液比>提取时间,其中提取温度和乙醇浓度有显著性差异(P < 0.05)。综合考虑,确定对萼猕猴桃苷E的最佳提取工艺为A1B2C2D2,即以对萼猕猴桃叶粉末25倍量的55%乙醇,95 ℃回流提取1 h。
表 1 L9(34)正交试验结果及直观分析(n = 3)
编号 A(%) B(w/V) C(t/h) D(T/ ℃) 对萼猕猴桃苷E含量(mg/g) 1 2 3 1 55(1) 1∶20(1) 0.5(1) 90(1) 7.94 7.98 7.87 2 55(1) 1∶25(2) 1(2) 95(2) 7.99 7.98 8.09 3 55(1) 1∶30(3) 1.5(3) 100(3) 7.89 7.85 7.98 4 65(2) 1∶20(1) 1(2) 100(3) 7.87 7.81 7.77 5 65(2) 1∶25(2) 1.5(3) 90(1) 7.85 7.99 7.97 6 65(2) 1∶30(3) 0.5(1) 95(2) 7.91 7.93 8.05 7 75(3) 1∶20(1) 1.5(3) 95(2) 7.91 7.91 7.81 8 75(3) 1∶25(2) 0.5(1) 100(3) 7.84 7.75 7.81 9 75(3) 1∶30(3) 1(2) 90(1) 7.88 7.97 7.87 K1 71.57 70.87 71.08 71.32 K2 71.15 71.27 71.23 71.58 K3 70.75 71.33 71.16 70.57 k1 7.95 7.87 7.90 7.92 k2 7.91 7.92 7.91 7.95 k3 7.86 7.93 7.91 7.84 R 0.09(2) 0.05(3) 0.02(4) 0.11(1) 表 2 对萼猕猴桃叶提取工艺方差分析
方差来源 离均差平方和 自由度 均方 F值 P值 A 0.037 2 0.019 4.989 0.019* B 0.014 2 0.007 1.856 0.185 C 0.001 2 0.001 0.167 0.847 D 0.061 2 0.031 8.161 0.003** 误差 0.067 18 0.004 合计 0.181 26 注:f1=2,f2=18,F0.05(2, 18)=3.55;*P<0.05,**P<0.01. -
根据优选出的提取方法制备3份对萼猕猴桃叶提取液,按“2.1.1”项下色谱条件进样测定,所测对萼猕猴桃叶中对萼猕猴桃苷E的提取率为99.32%,得率为0.80%,RSD均小于1%,表明所选工艺稳定,可用于对萼猕猴桃叶中对萼猕猴桃苷E的提取。
-
(1)大孔树脂预处理
取5种型号的大孔树脂D101、AB-8、DM130、HPD100、NKA-9适量,置于250 ml锥形瓶内,倒入高于树脂层3 cm的95%乙醇,置摇床振摇12 h,继续用95%乙醇洗涤,直至洗涤液不浑浊。然后加入蒸馏水至摇床振摇清洗,直至无醇味。
(2)大孔树脂型号的筛选
精密称取已处理的上述树脂各0.5 g,置于50 ml具塞锥形瓶中,分别加入最佳提取工艺制备的对萼猕猴桃叶浓缩提取液10 ml,盖紧瓶塞,置于恒温振荡器中振摇12 h(150 r/min),达到饱和吸附后过滤,收集未吸附溶液,测定未吸附液中对萼猕猴桃苷E的含量,计算各树脂的吸附率。将上述静态吸附过的树脂用吸水纸吸干溶液,各用10 ml 75%乙醇溶液解吸附,相同条件下振摇,收集解吸附液,测定溶液中对萼猕猴桃苷E的含量,计算各树脂的解吸附率。结果见表3,综合吸附和解吸附性能考虑,最终根据权重大小选择D101型大孔树脂进行后续实验。
表 3 不同型号大孔树脂对对萼猕猴桃苷E静态吸附和解吸附的测定结果
树脂型号 吸附率(%) 解吸率(%) 权重(%) D101 91.54 96.67 88.49 AB-8 88.71 98.01 86.94 DM130 62.10 89.80 55.76 HPD100 87.82 97.25 85.41 NKA-9 87.15 97.64 85.09 (3)上样液浓度考察
称取5份已处理的D101大孔树脂20 g,湿法装柱,分别加入浓度为0.07、0.16、0.24、0.33、0.55 mg/ml的对萼猕猴桃叶提取液5 ml,完全吸附后加3 BV水除杂,3 BV 95%乙醇洗脱,收集洗脱液,测定洗脱液中对萼猕猴桃苷E的量。结果表明,对萼猕猴桃苷E的转移率随上样液浓度的增加而升高(图3A),当上样液浓度大于0.55 mg/ml时,其流动性变差,不予考虑,因此选择0.55 mg/ml作为合适的上样液浓度。
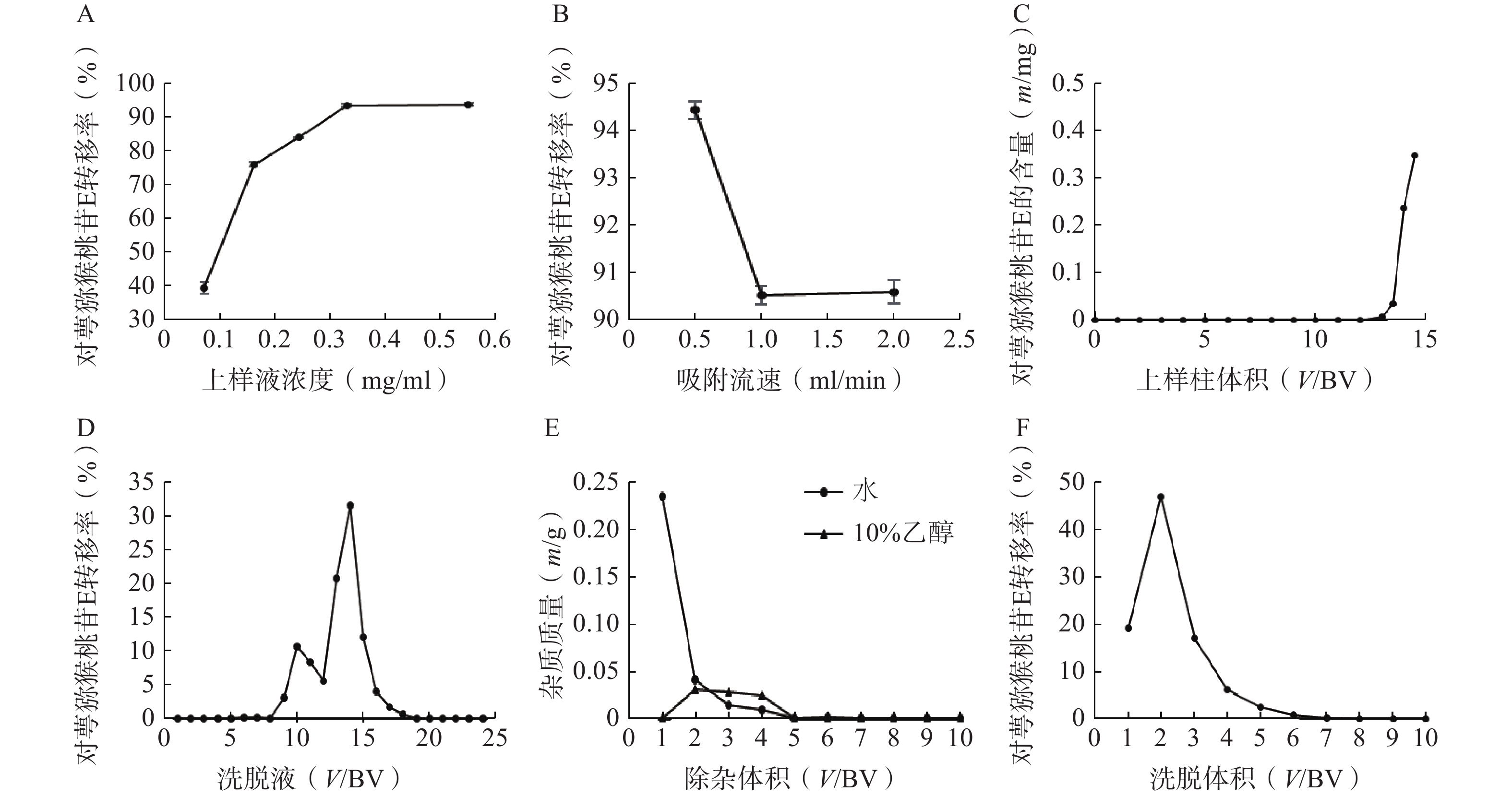
图 3 D101型大孔树脂纯化对萼猕猴桃叶提取液工艺考察结果
(4)吸附流速考察
称取3份已处理的D101大孔树脂20 g,湿法装柱,量取浓度为0.55 mg/ml的上样液5 ml,分别以0.5、1、2 ml/min的流速上柱吸附,完全吸附后加3 BV水除杂,3 BV 95%乙醇洗脱,收集洗脱液,测定对萼猕猴桃苷E的量。结果表明,吸附流速增大,对萼猕猴桃苷E的转移率下降(图3B),故选择0.5 ml/min作为合适的吸附流速。
(5)最大上样体积考察
称取已处理的D101大孔树脂20 g(1 BV=30 ml),湿法装柱,制备一定体积的浓度为0.55 mg/ml的上样液,吸附流速为0.5 ml/min,收集流出液(每15 ml收集1管),根据泄露曲线确定最大上样量。结果表明,当上样体积大于12 BV时,流出液中检测到对萼猕猴桃苷E,且泄露量逐渐增大(图3C),因此确定上样量为12 BV。
(6)梯度洗脱曲线考察
称取已处理的D101大孔树脂20 g,湿法装柱,量取浓度为0.55 mg/ml的上样液12 BV,吸附流速为0.5 ml/min,完全吸附后,加水和10%、30%、50%、70%、95%乙醇各4 BV依次洗脱,收集各段洗脱液,每1 BV为1份。结果显示,水和10%乙醇洗脱液中有极少量的对萼猕猴桃苷E,其主要集中在30%和50%乙醇洗脱液中(图3D),故选择水和10%乙醇除杂,50%乙醇洗脱。
(7)除杂溶剂用量考察
称取已处理的D101大孔树脂20 g,湿法装柱,量取浓度为0.55 mg/ml的上样液12 BV,以0.5 ml/min的流速上样吸附,加水和10%乙醇除杂,均冲洗10 BV,按柱体积收集各段除杂液,将其烘干称重。结果表明,水和10%乙醇除杂液烘干质量均在第5 BV后无明显变化(图3E),因此除杂体积均控制在5 BV。
(8)洗脱溶剂用量考察
称取已处理的D101大孔树脂20 g,湿法装柱,量取浓度为0.55 mg/ml的上样液12 BV,吸附流速为0.5 ml/min,依次加水和10%乙醇各5 BV除杂,50%乙醇洗脱10 BV,收集洗脱液,测定洗脱液中对萼猕猴桃苷E的量。结果表明,7 BV后洗脱液中对萼猕猴桃苷E的量基本可以忽略(图3F),因此选择50%乙醇洗脱液的用量为7 BV。
(9)D101型大孔树脂柱纯化对萼猕猴桃苷E工艺验证
称取已处理的D101型大孔树脂20 g,湿法装柱,量取浓度为0.55 mg/ml的上样液12 BV,以0.5 ml/min的流速进行动态吸附,完全吸附后,依次加水和10%乙醇各5 BV除杂,50%乙醇7 BV洗脱,除杂和洗脱流速均为2 ml/min,收集洗脱液,测定对萼猕猴桃苷E的含量,计算该步骤中对萼猕猴桃苷E的转移率,并将洗脱液浓缩干燥至恒重,备用。结果表明,在大孔树脂纯化步骤中,对萼猕猴桃叶中对萼猕猴桃苷E的转移率为91.36%。该纯化工艺稳定可行,可用于对萼猕猴桃叶提取液的纯化。
-
(1)硅胶柱装填
称取200~300目硅胶适量,用初始比例的洗脱液充分溶胀后湿法装柱,尽可能地排出柱子中的气泡,待其沉降后加入洗脱液充分冲洗压实,直至硅胶层界面不再下降。收集D101型大孔树脂50%乙醇洗脱部分所得的干燥样品,适量甲醇溶解后与100~200目硅胶按质量比1∶1~1∶1.5于50 ℃水浴锅上充分混匀蒸干,干法上样后在其表面加一层空白硅胶,以不同比例的洗脱液洗脱,收集各段洗脱液,测定对萼猕猴桃苷E的含量。
(2)不同洗脱系统的考察
①二氯甲烷-甲醇洗脱系统的考察:采用湿法装柱干法上样的方式进行硅胶柱色谱,依次用二氯甲烷-甲醇15∶1、10∶1、8∶1、5∶1、3∶1、1∶1、1∶5、0∶1洗脱,每个梯度洗脱5 BV,收集各段洗脱液,测定对萼猕猴桃苷E的含量。结果表明,对萼猕猴桃苷E主要集中在10∶1和8∶1洗脱部位(图4A),可选择二氯甲烷-甲醇8∶1洗脱5 BV进行硅胶柱色谱。
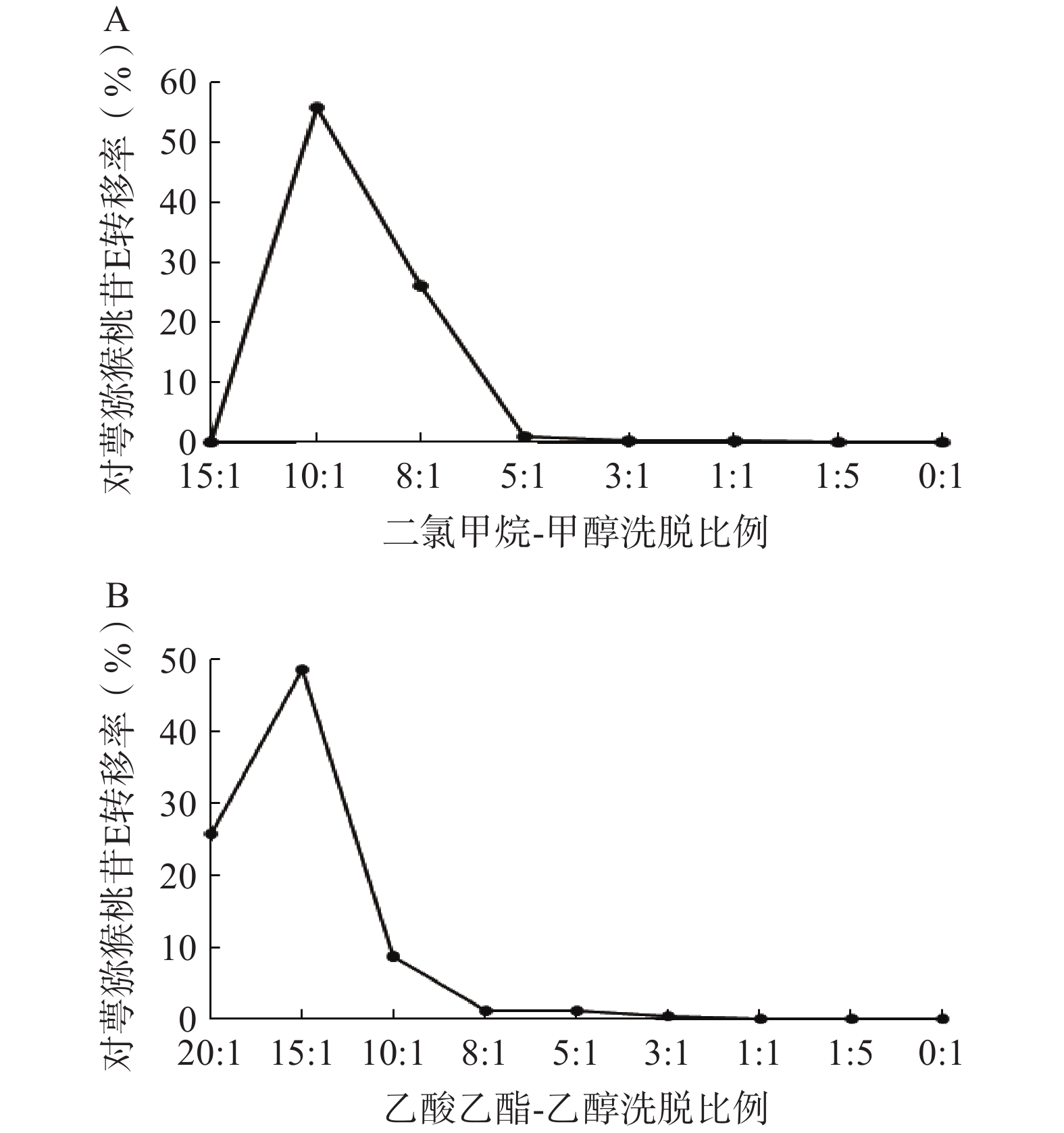
图 4 硅胶柱色谱不同洗脱系统考察结果
②乙酸乙酯-乙醇洗脱系统的考察:依次用乙酸乙酯-乙醇20∶1、15∶1、10∶1、8∶1、5∶1、3∶1、1∶1、1∶5、0∶1洗脱,其余步骤同上。结果显示,对萼猕猴桃苷E在10∶1条件下基本已完全洗脱(图4B),因此可选择乙酸乙酯-乙醇洗脱系统10∶1洗脱5 BV。
在硅胶柱色谱实验中,使用上述两种洗脱体系所得对萼猕猴桃苷E的转移率分别为82.09%、83.21%,两者相差不大,考虑到洗脱溶剂的毒性安全性问题,选择乙酸乙酯-乙醇系统作为硅胶柱色谱步骤的洗脱体系。
(3)硅胶柱纯化对萼猕猴桃苷E工艺验证
硅胶柱色谱采用湿法装柱干法上样的方式进行操作,上样后以乙酸乙酯-乙醇10∶1洗脱5 BV,收集洗脱液,测定洗脱液中对萼猕猴桃苷E的含量,得该步骤对萼猕猴桃苷E的转移率为83.52%。
-
将浸泡在甲醇中的ODS填料倒入色谱柱中,待其沉降完全后以初始洗脱比例30%甲醇溶液置换柱子中的纯甲醇溶液。将硅胶柱色谱所得的洗脱液浓缩干燥后用少量甲醇溶解过滤膜后进行湿法上样,待样品完全吸附后,依次用30%、40%、50%甲醇各洗脱2 BV,分段收集50%甲醇洗脱部分,通过液相色谱结果进行合并,合并液再次以相同条件进行ODS柱色谱,最终得到较纯的对萼猕猴桃苷E单体化合物(图5),该步骤中对萼猕猴桃苷E的转移率为70.37%,其纯度为99.9%,表明该方法分离纯化效果较好。
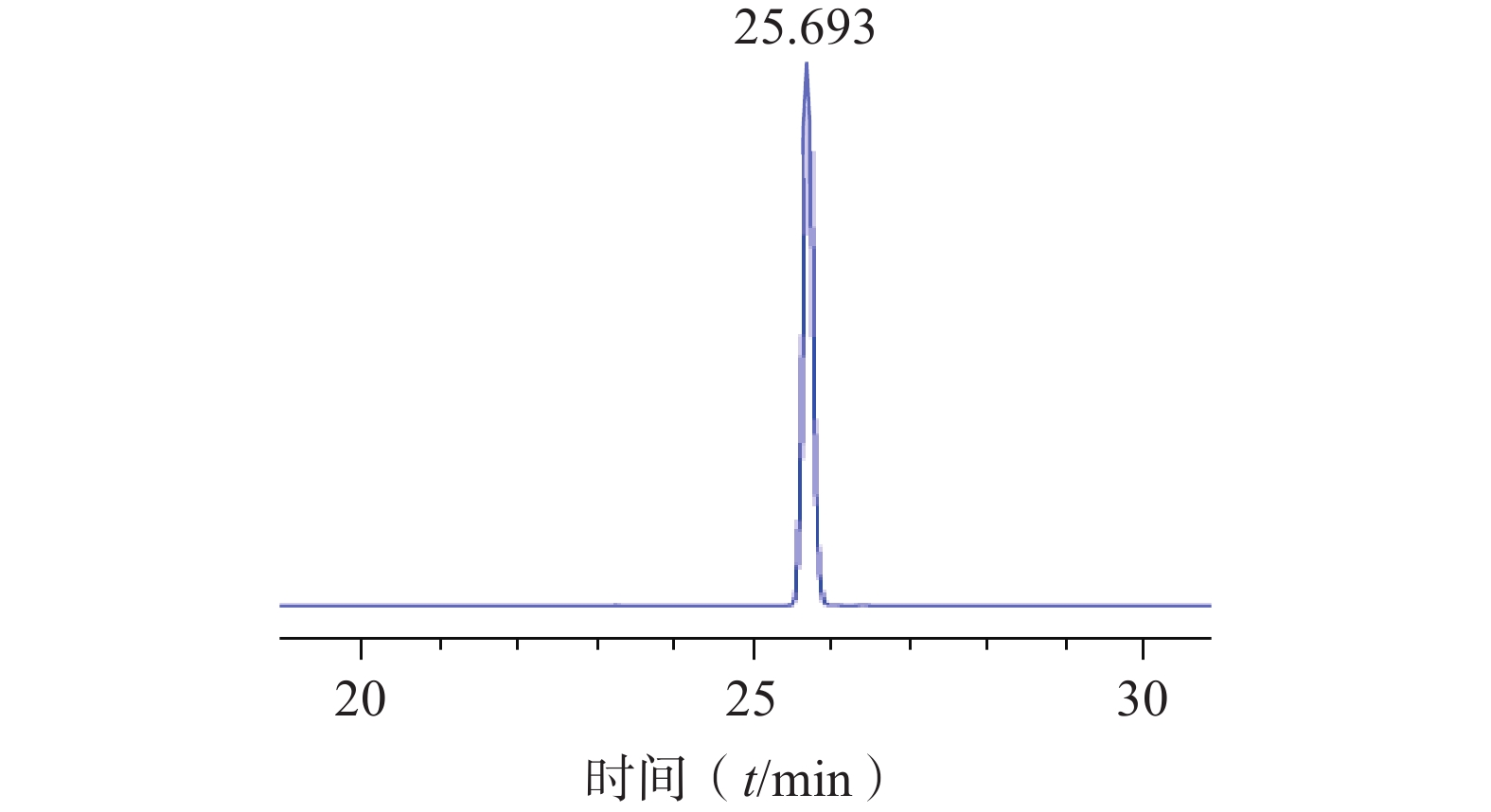
图 5 ODS柱色谱所得对萼猕猴桃苷E的HPLC图
-
按照优选的对萼猕猴桃苷E提取分离纯化方法对其工艺进行验证。称取对萼猕猴桃叶粉末25 g,以25倍量55%乙醇95 ℃回流提取1 h,滤液减压浓缩。将已处理的D101型大孔树脂进行湿法装柱,量取浓度为0.55 mg/ml的上样液12 BV,吸附流速为0.5 ml/min,依次加水和10%乙醇各5 BV除杂,50%乙醇7 BV洗脱,洗脱液浓缩干燥。硅胶柱采用湿法装柱干法上样的方式进行柱色谱,上样后以乙酸乙酯-乙醇10:1洗脱5 BV,收集并浓缩干燥洗脱液。ODS柱经上样后依次用30%、40%、50%甲醇各洗脱2 BV,分段收集50%甲醇洗脱部分,较纯部分合并后再次以相同条件进行ODS柱色谱,最终得到黄色粉末状物质,HPLC进样检测,与对照品比对后确定该物质为对萼猕猴桃苷E。该提取分离纯化工艺所得对萼猕猴桃苷E的转移率为53.70%,得率为0.35%,纯度为99.9%,表明该方法稳定可行,可用于对萼猕猴桃叶中对萼猕猴桃苷E的提取分离纯化。
-
中药的提取方法包括热回流提取、水煎煮提取、超声辅助提取等方法[8],在前期提取方法及提取试剂的考察中,我们使用了水及不同浓度的乙醇考察提取溶剂对对萼猕猴桃叶中对萼猕猴桃苷E提取率的影响,结果表明,乙醇浓度在60%左右时其提取率最大。此外,通过对超声提取法、热回流提取法及渗漉法三种提取方法的比较,发现热回流提取法具有时间较短、节省溶剂、提取率较大的优势,因此在本实验中选用乙醇回流提取法进行单因素考察和正交试验。
在回流提取实验的单因素考察中,除了乙醇浓度、料液比、提取时间和提取温度外,提取次数也是一个重要的考察因素。本实验发现,第2次回流提取所得的提取液中对萼猕猴桃苷E含量极低,基本可以忽略,因此在实验中确定提取次数为1次。
大孔吸附树脂柱色谱是分离纯化黄酮类化合物常用的方法之一[9],其吸附与解吸附性能对化合物的转移率有很大影响。本实验中,我们对D101型大孔树脂进行静态吸附与解吸附动力学考察,分别在不同时间点吸取未吸附液及解吸附液,测定对萼猕猴桃苷E的含量。结果显示,D101型大孔树脂属于慢速吸附型树脂,在上样时,应尽量降低流速,增加样品与树脂的接触时间,其解吸附能力较强,洗脱时可加大流速。
葡聚糖凝胶柱和ODS柱常作为化合物精制纯化的关键一步。前者主要是依据分子筛原理,由于化合物的分子量不同,在凝胶柱中受到的阻滞作用也不同,导致分子量大的化合物先被洗脱下来[10];后者是根据化合物极性的不同进行分离的,由于反相柱中固定相极性小于流动相极性,致使极性较大的化合物先从柱中流出。在硅胶柱色谱后,我们发现对萼猕猴桃叶洗脱液中主要成分为两个黄酮苷类化合物,两者结构式仅有一个乙酰基之差,使用葡聚糖凝胶柱始终无法完全分离。其中对萼猕猴桃苷E的极性与另一黄酮类化合物相比极性较小,通过高效液相色谱可以将两个化合物分离,且分离度较好。因此,选用ODS反相柱进行纯化,最终得到对萼猕猴桃苷E单体化合物。
本实验从中药资源非药用部位的开发利用角度出发,对对萼猕猴桃叶进行研究,首次设计出一条稳定可行、重复性好的提取分离纯化工艺从对萼猕猴桃叶中分离得到对萼猕猴桃苷E,整个工艺操作简易,提取效率高,所得对萼猕猴桃苷E纯度为99.9%。作为一个新化合物,其药物开发过程有很长的路要走,本实验所做的提取分离纯化工艺研究为对萼猕猴桃苷E进一步的药代动力学研究、安全性药理研究等内容的实施奠定了物质基础。
Study on the extraction, separation and purification process of actinoside E
-
摘要:
目的 优选对萼猕猴桃苷E的提取分离纯化工艺。 方法 以对萼猕猴桃苷E的含量为指标,采用单因素考察结合正交试验确定对萼猕猴桃苷E的最佳提取工艺;通过优化大孔吸附树脂柱、硅胶柱和ODS柱的色谱条件,对提取物进行分离纯化。 结果 对萼猕猴桃苷E的最佳提取工艺为:以25倍量的55%乙醇95 ℃加热回流1 h。优化的分离纯化工艺为:D101型大孔树脂柱以50%乙醇洗脱7 BV;硅胶柱以乙酸乙酯-乙醇10∶1洗脱5 BV;ODS柱重复纯化50%甲醇洗脱部分,最终得到对萼猕猴桃苷E单体化合物。整个工艺对萼猕猴桃苷E的转移率为53.70%,得率为0.35%,纯度达99.9%。 结论 该工艺稳定可行,可为对萼猕猴桃苷E的开发利用提供物质基础。 Abstract:Objective To optimize the extraction, separation and purification process of actinoside E. Methods Single factor experiment combined with orthogonal test was used to determine the optimal extraction process of actinoside E using its content as an index. The extracts were separated and purified by optimizing the chromatographic conditions of macroporous resin, silica gel and ODS column. Results 25 times amount of 55% ethanol with heating reflux at 95 ℃ for one hour were used as the optimal extraction process of actinoside E. The optimum separation and purification process was as follows: D101 macroporous resin column was eluted with 7 BV of 50% ethanol, silica gel column was eluted with 5 BV of ethyl acetate-ethanol(10∶1)and 50% methanol eluted fraction was purified repeatedly by ODS column to obtain actinoside E. The transfer rate of actinoside E in the whole process was 53.70%, the yield was 0.35%, and the purity was 99.9%. Conclusion The process is stable and viable, which can provide material foundation for the development and utilization of actinoside E. -
Key words:
- actinoside E /
- extraction /
- separation and purification /
- chromatography
-
STAT3是一种重要的转录因子,参与众多细胞因子和生长因子受体的信号转导,在细胞生长和细胞凋亡等多种细胞过程中发挥着关键作用[1-2]。STAT3的活化可以通过刺激白介素-6受体(IL-6R)、Janus酪氨酸激酶、BCR-ABL和SRC家族激酶等来启动[3]。STAT3经磷酸化活化后形成同源和异源二聚体,并易位至细胞核,发挥转录激活因子的作用[4-6]。目前,越来越多的证据显示,多种恶性肿瘤存在STAT3的过度激活,包括前列腺癌、肺癌、乳腺癌、皮肤癌和宫颈癌等,抑制STAT3的磷酸化成为一种很有前景的治疗策略。此外,STAT3还与肝损伤、纤维化、风湿性关节炎、心肌缺血等疾病有关[7]。尽管一些STAT3抑制剂正在进行临床试验,但迄今为止尚未批准STAT3抑制剂用于癌症的治疗。因此,仍然迫切需要发现潜在的STAT3抑制剂[8]。
SPR是一种光学生物传感技术,该技术利用光学测量的折射率变化,分析样品与固定在SPR传感器上的分子的结合情况。因其无需标记样品,具有高灵敏度,能实时检测生物分子间的相互作用而被广泛运用于医疗检测、药物筛选、环境监测和食品检测等领域[9]。
本课题采用SPR技术从中药单体中筛选能与STAT3特异性结合的小分子化合物,通过蛋白免疫印迹技术和双荧光素酶报告基因考察小分子对STAT3的抑制作用,采用分子对接技术拟合化合物与STAT3的结合模式,明确其可能的作用位点,从而为STAT3抑制剂的发现提供理论指导和实践经验。
1. 材料与方法
1.1 实验材料
1.1.1 实验试剂
DMSO(美国Sigma公司);EDC/NHS(GE公司);胰酶(美国Gibco公司);DMEM培养基(美国Corning公司);胎牛血清(美国Gibco公司);细胞裂解液、PMSF、30%丙烯酰胺溶液、1.5 mol/L Tris(pH=8.8)、1.0 mol/L Tris(pH=6.8)、10%SDS、TEMED、BCA试剂盒、双荧光素酶报告基因检测试剂盒(上海碧云天生物技术有限公司);硝酸纤维素膜(德国Sartorius Stedim公司);转染试剂(美国Life Technology公司)
1.1.2 实验仪器
Biacore T2000(GE医疗生命科学公司);电子天平(上海天平仪器厂);电泳仪(美国Bio-Rad公司);制冰机(德国Eppendorf公司);−80℃低温冰箱(美国Thermo公司);CO2细胞培养箱(美国Thermo公司);离心管(美国Corning公司);低温高速台式离心机(美国Thermo公司);移液枪(德国Eppendorf公司);超纯水仪(美国Millipore公司);多功能酶标仪(美国Thermo公司)。
1.1.3 实验细胞
HeLa细胞(购自上海碧云天生物科技有限公司,由本实验室冻存、培养);HeLa-STAT3-Luc细胞(由本实验室构建、培养、冻存)。
1.2 实验方法
1.2.1 Biacore鉴定STAT3特异性结合小分子
(1)STAT3预富集
将STAT3纯化蛋白用去离子水溶解并配成1 g/L的蛋白母液,用4种不同pH的醋酸盐缓冲液(pH4.0、pH4.5、pH5.0、pH5.5)稀释蛋白母液至50 mg/L,进样,于Biacore预富集系统检测不同pH条件下蛋白STAT3的响应值,确定最佳蛋白偶联条件。
(2) STAT3偶联
在预富集实验中所得最佳pH条件下,将STAT3稀释至50 mg/L,通过EDC/NHS活化CM5芯片表面羧基,然后通过羧基氨基缩合反应将STAT3键合到CM5芯片上,乙醇胺封闭未结合的羧基,从而实现STAT3偶联到CM5芯片上的目的。
(3)亲和力分子
将中药小分子单体化合物用DMSO溶解,然后用PBS稀释成32 μmol/L (DMSO终浓度为5%)的样品后进样,流动相为5%DMSO的PBS溶液,通过Biacore系统分析其流过STAT3蛋白表面的响应值,筛选出对STAT3响应值较高(高于阳性对照或与其相当)的单体化合物作为候选化合物。
(4)动力学分析
将候选化合物浓度以二倍比进行梯度稀释,浓度范围为0.0625~64 μmol/L(DMSO终浓度均为5%),通过Biacore系统分析获得结合响应值,根据响应值与候选化合物浓度之间的量效关系绘制动力学曲线,根据曲线拟合情况判断候选化合物与STAT3的结合特异性,从而找到能与STAT3蛋白特异性结合的小分子单体。
1.2.2 蛋白免疫印迹(Western-blot)检测化合物对STAT3磷酸化的抑制情况
HeLa细胞以5×105个/孔接种于6孔板,于37 ℃、5%CO2细胞培养箱中培养过夜。次日,加入不同浓度的化合物,作用24 h后,加入100 μl Western及IP细胞裂解液(含1 mmol/L PMSF),冰上裂解25 min,收集蛋白于1.5 ml离心管,12 000 r/min,4 ℃离心10 min,吸取上清液,使用BCA蛋白定量试剂盒进行总蛋白定量。蛋白样品中加入5×蛋白上样缓冲液,煮沸5 min,进行SDS-PAGE凝胶电泳。电泳结束后,在250 mA恒流电下将蛋白从凝胶转移到NC膜上,5%脱脂牛奶封闭,进行一抗(p-STAT3/STAT3)、二抗孵育,结束后在红外双色激光成像系统(Odyssey)上扫膜检测700和800通道激发的荧光信号,观察各泳道中蛋白表达情况。
1.3 双荧光素酶报告基因鉴定化合物抑制STAT3磷酸化的作用
HeLa-STAT3-Luc细胞计数后按1×105个/孔接种于24孔板,置于37 ℃、5%CO2细胞培养箱中培养24 h,加入不同浓度化合物孵育4 h,然后加入IL-6(100 ng/ml)和IL-6R(100 ng/ml)共同刺激24 h,弃上清液,每孔加入120 μl细胞裂解液,离心后取5 μl上清液转移至新的384孔板,每孔加入25 μl荧光素酶1液,使用多功能酶标仪检测荧光值,然后加入25 μl荧光素酶2液,再次测荧光值,定内参。
1.4 分子对接技术
分子对接以Protein Preparation Wizard模块处理蛋白,选择STAT3蛋白与小分子抑制剂的晶体复合物6NUQ,依次去水、加氢,以LigPrep模块处理配体,力场优化均采用OPLS2005模式,其余参数均使用默认;以Grid模块建立蛋白对接坐标,范德华力半径设置为1.0;采用精确对接模式(XP)的方法进行对接,对接结果用PyMol软件作图。
1.5 统计学分析
数据以(x±s)表示,采用统计学软件SPSS19.0对数据进行单因素方差分析,进行组间差异比较。P<0.05认为差异具有统计学意义。
2. 实验结果
2.1 Biacore系统预富集STAT3蛋白
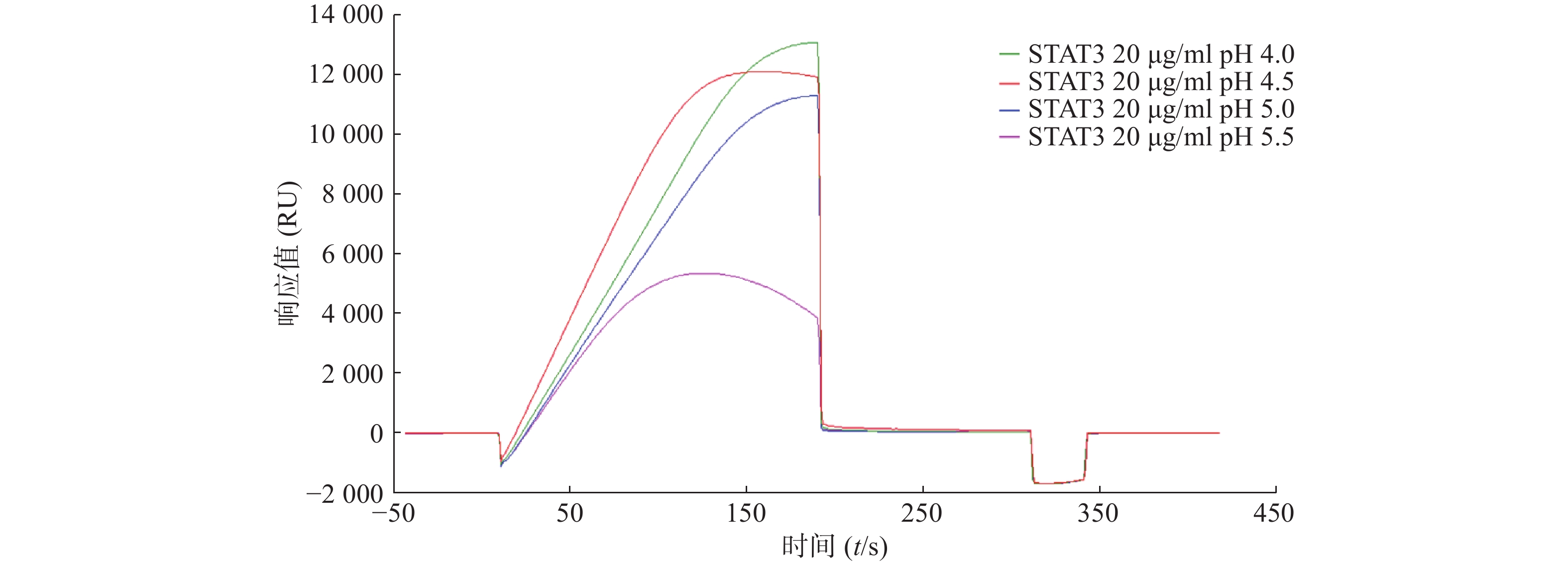
为了研究STAT3蛋白的最佳偶联条件,采用不同pH的醋酸盐缓冲液稀释蛋白,进行预富集分析,结果显示STAT3在pH4.0的条件下响应值最高(图1)。因此,后续实验选择pH4.0的缓冲液进行偶联。
2.2 Biacore系统偶联STAT3蛋白
取市售的STAT3蛋白用pH4.0醋酸盐缓冲液稀释至50 mg/L,通过Biacore系统的内置程序偶联到CM5芯片上,结果显示STAT3偶联量为8 000 RU,达到预计偶联水平(图2)。
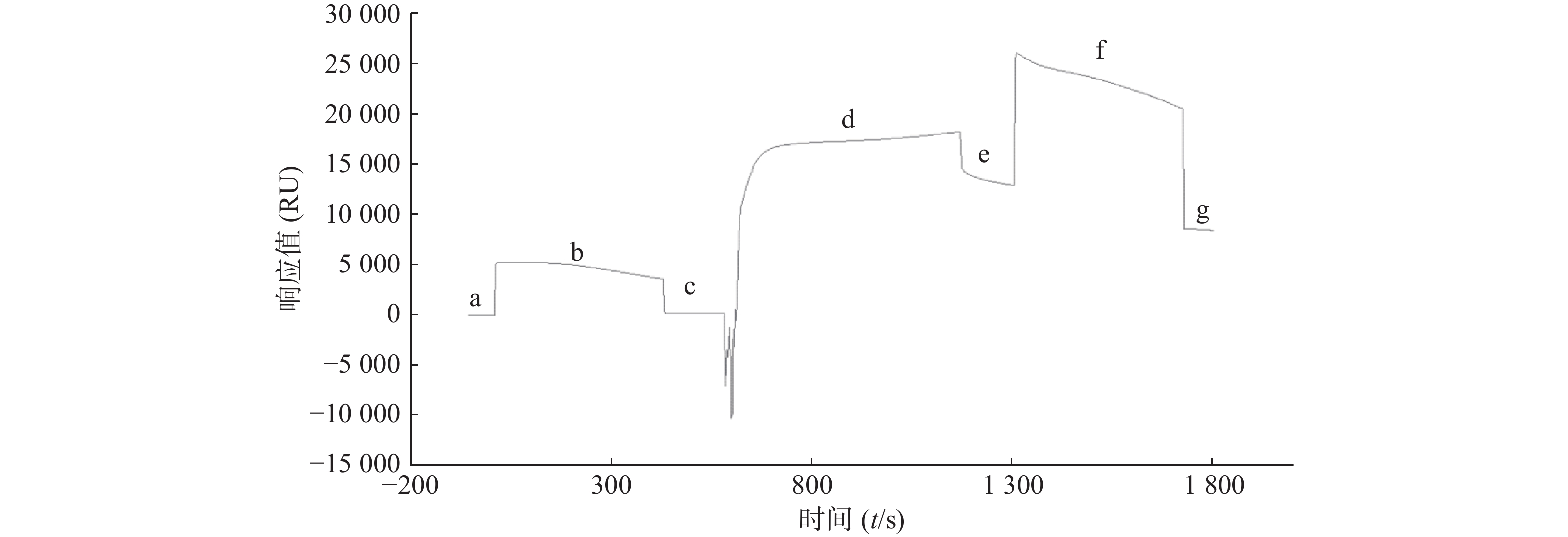 图 2 Biacore系统STAT3蛋白偶联情况注:a段表示流动相基线水平稳定;b段表示EDC/NHS活化CM5芯片表面的过程;c段为流动相清洗活化试剂;d段为STAT3蛋白固定到芯片表面的过程;e段代表流动相清洗未结合到芯片表面而残留的STAT3蛋白;f段代表乙醇胺封闭芯片表面多余羧基过程;g段为流动相清洗剩余乙醇胺的过程。
图 2 Biacore系统STAT3蛋白偶联情况注:a段表示流动相基线水平稳定;b段表示EDC/NHS活化CM5芯片表面的过程;c段为流动相清洗活化试剂;d段为STAT3蛋白固定到芯片表面的过程;e段代表流动相清洗未结合到芯片表面而残留的STAT3蛋白;f段代表乙醇胺封闭芯片表面多余羧基过程;g段为流动相清洗剩余乙醇胺的过程。2.3 筛选中药单体小分子化合物
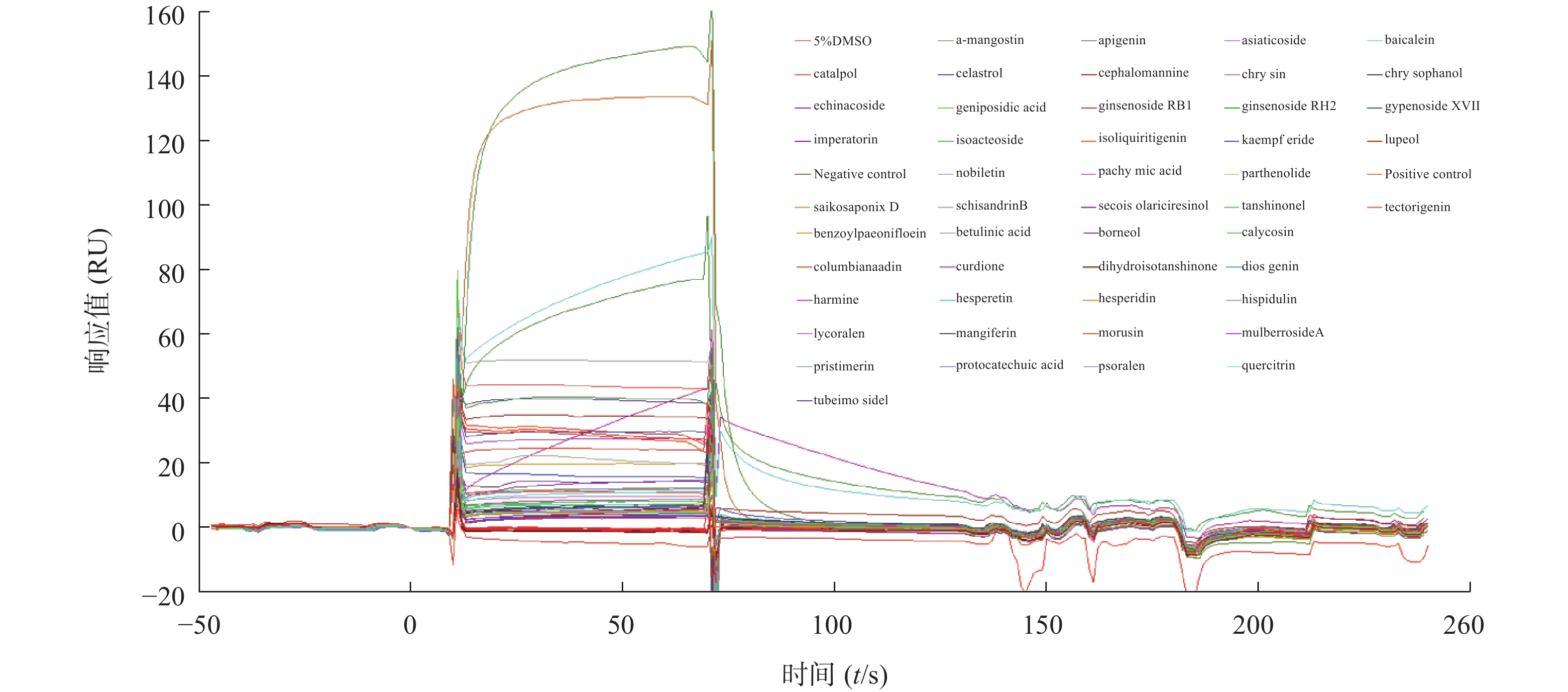
为了筛选能够结合STAT3蛋白的小分子化合物,我们将50种中药单体统一稀释到32 μmol/L,利用Biacore系统检测结合情况,通过响应值观察小分子与STAT3蛋白的结合强度。结果发现,不同小分子化合物与STAT3蛋白的结合存在差异(图3),我们以阳性对照(C188-9)为标准,筛选响应值不低于阳性对照响应值的化合物,得到了梓醇(catalpol)、黄芩素(baicalein)、芹黄素(apigenin)、槲皮素(quercitrin)、人参皂苷(ginsenoside)、京尼平苷酸(geniposidic acid)、桑辛素(morusin)等10多种小分子作为候选化合物,接着进行动力学分析以确定它们与STAT3蛋白的结合特异性。
2.4 候选化合物的动力学分析
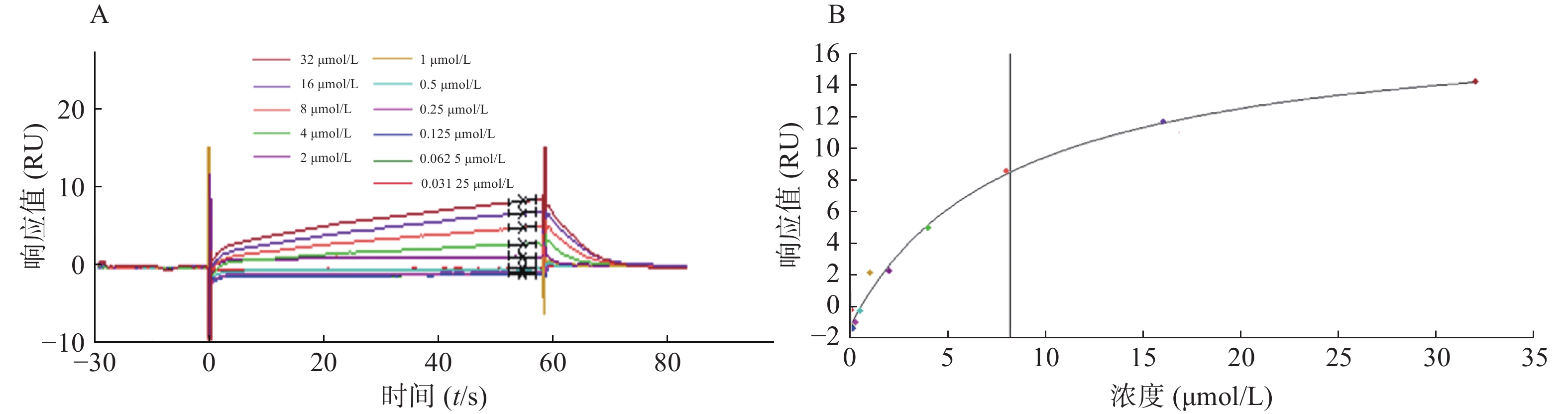
为了验证候选化合物与STAT3蛋白的结合是否为特异性结合,我们将候选化合物进行梯度稀释,通过Biacore系统分析获得结合响应值,绘制动力学结合曲线。对10多种化合物均进行动力学分析,发现只有芹黄素与STAT3的结合具有特异性。芹黄素与STAT3的结合响应值随着药物浓度的增大而升高,当浓度增大到一定值时响应值呈水平趋势不再变化,说明高浓度芹黄素同STAT3的结合存在饱和现象,即芹黄素与STAT3的结合为特异性结合(图4)。因此,选择芹黄素作为可能的STAT3抑制剂进行生物学验证。
2.5 Western-blot检测结果
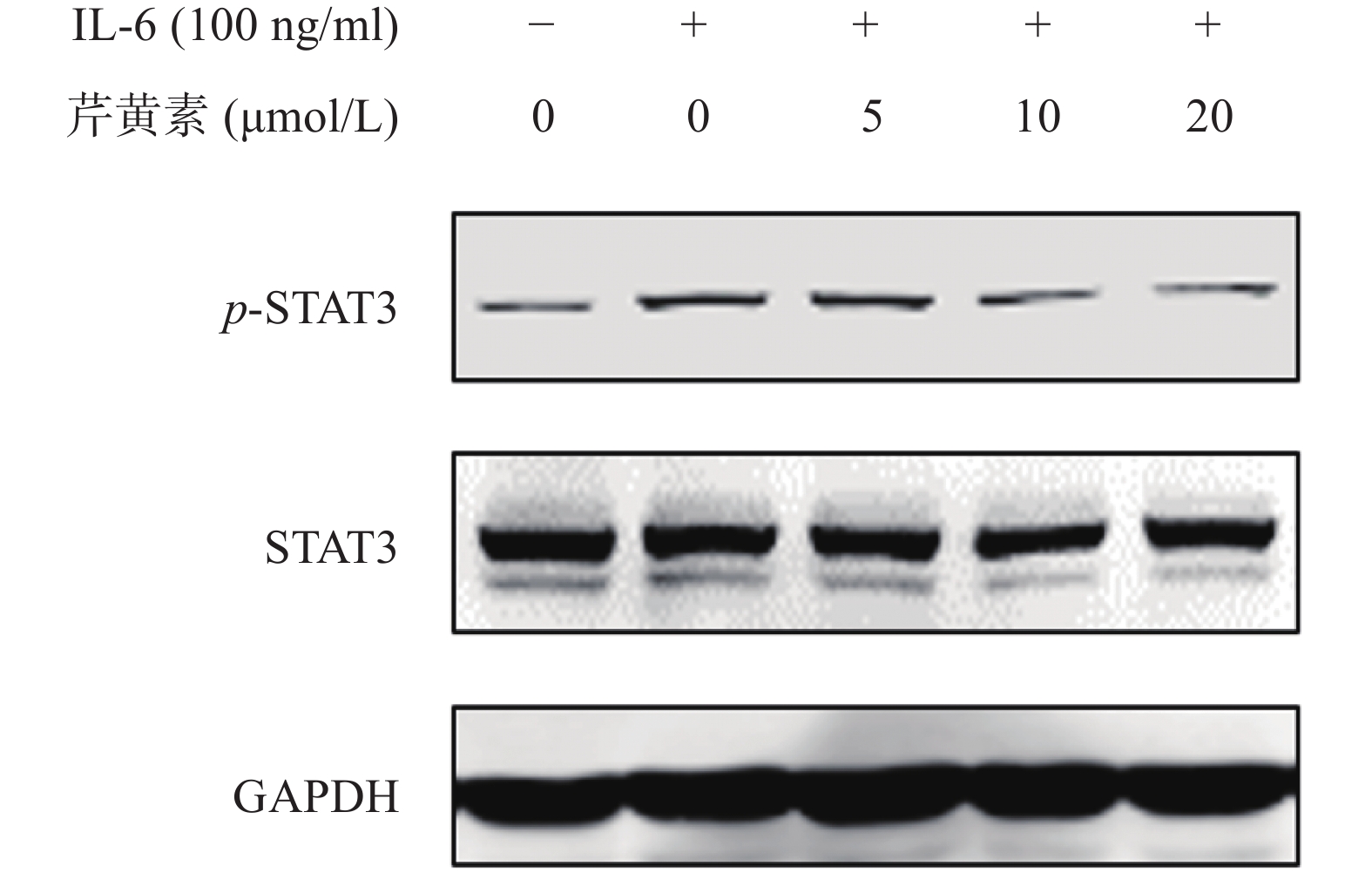
动力学分析结果证实芹黄素可以特异性结合STAT3,为了确证芹黄素对STAT3磷酸化的抑制作用,我们利用IL-6诱导活化STAT3,通过Western-blot检测芹黄素对STAT3磷酸化的抑制作用。结果发现,IL-6可以显著刺激HeLa细胞STAT3的活化,而芹黄素能剂量依赖地抑制IL-6诱导的STAT3磷酸化,表明芹黄素可能是STAT3的抑制剂(图5)。
2.6 双荧光素酶报告基因检测结果
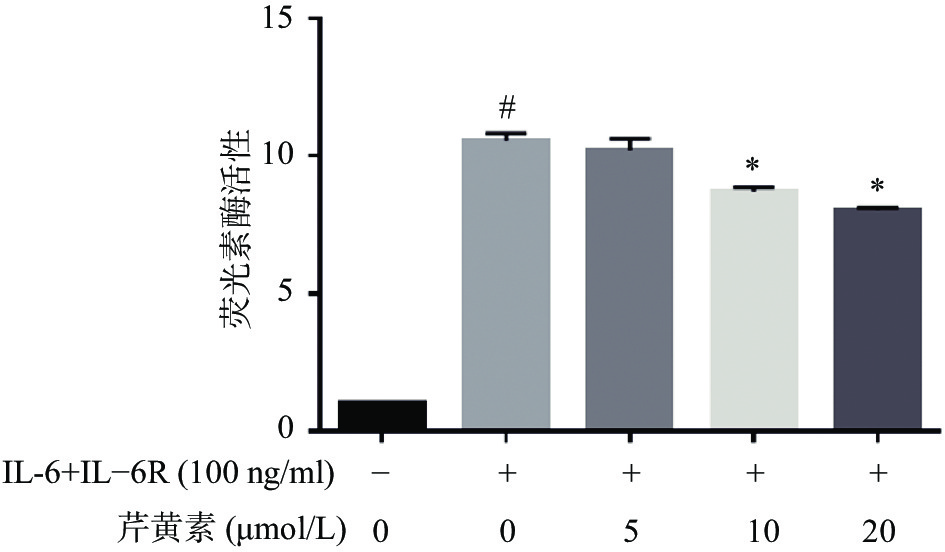
为了进一步确证芹黄素对STAT3的抑制作用,我们采用双荧光素酶报告基因系统研究芹黄素对STAT3转录活性的影响。结果显示,IL-6刺激可以显著促进STAT3的转录活性,10μmol/L、20 μmol/L芹黄素能够抑制IL-6诱导的STAT3转录活性的增加(图6)。以上结果表明芹黄素能够抑制STAT3的转录活性,进一步证实芹黄素是STAT3的小分子抑制剂。
2.7 分子对接结果
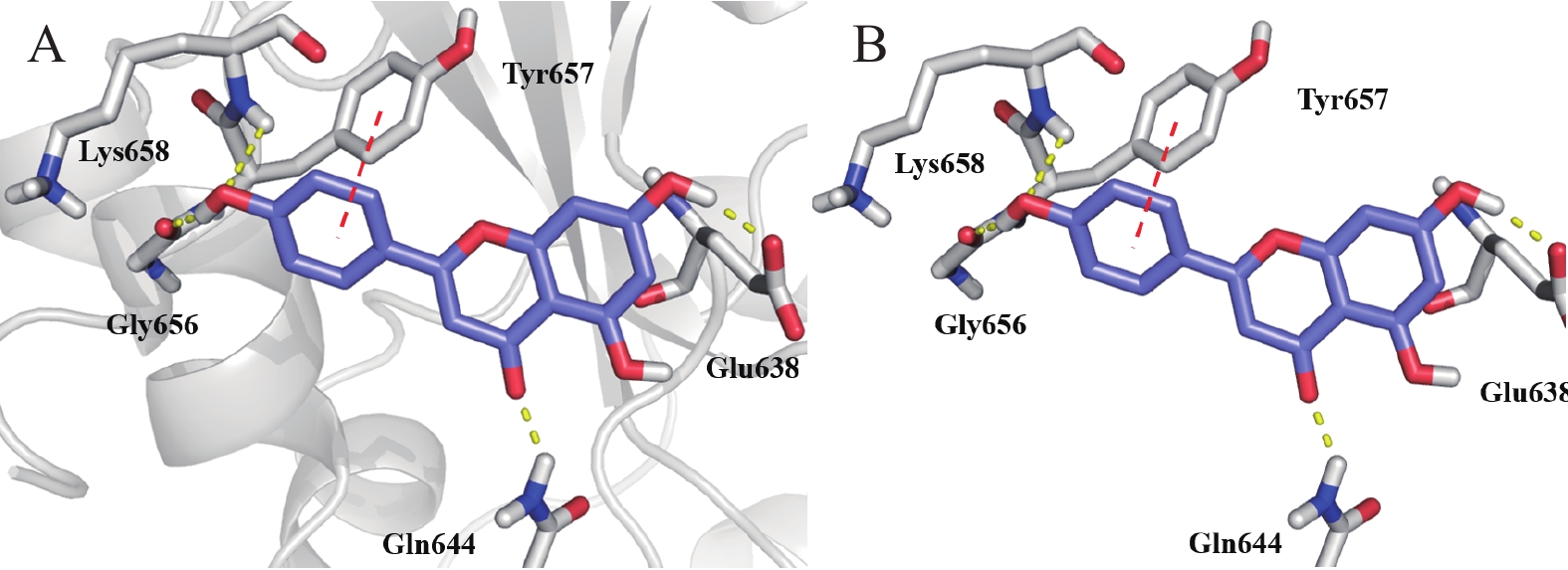
为了明确STAT3与芹黄素的相互作用情况,我们采用分子对接技术分析STAT3与芹黄素的结合位点。结果显示,芹黄素结合于STAT3蛋白的SH2结构域,占据了STAT3磷酸络氨酸的结合口袋。与关键残基Glu638、Gln644、Gly656、Lys658形成氢键相互作用,与Tyr657残基形成π-π相互作用(图7)。
3. 讨论
STAT3在肿瘤中的持续激活和过度表达与肿瘤细胞的多种恶性生物学特征密切相关。STAT3的活化受多种细胞因子、生长因子,生长因子受体,非受体蛋白酪氨酸激酶等多重信号分子的调控,目前已成为肿瘤治疗领域的研究热点之一[10]。 STAT3存在6个结构域,包括氨基末端结构域(NTD)、卷曲螺旋结构域(CCD)、DNA结合结构域(DBD)、接头结构域、Src同源结构域(SH2)和羧基末端反式激活(TAD)结构域。目前,对STAT3的直接抑制作用可以通过破坏SH2、DBD或NTD结构域来阻止功能性STAT3二聚体的形成。STAT3的直接抑制剂主要分为三类:肽,小分子抑制剂和寡核苷酸。间接抑制剂则通过靶向STAT3信号通路阻断上游信号通路(如IL-6和JAK通路)间接抑制STAT3[11-12]。目前,STAT3抑制剂的研发已成为肿瘤治疗领域的研究热点之一。
芹黄素是一种天然黄酮类化合物,具有抗炎、抗氧化和抗癌作用 [13],其在多种癌症中(如乳腺癌、肺癌、肝癌、前列腺癌等)表现出对细胞的生长抑制与促凋亡作用。芹黄素不仅能够通过内源性与外源性凋亡途径促进细胞凋亡,也能通过降低基质金属蛋白酶-2,-9的表达抑制肿瘤细胞侵袭[14]。但是,芹黄素与STAT3的关系尚未研究,其对STAT3的抑制作用也没有报道。
本研究利用表面等离子体共振技术,从50个中药单体中筛选出能与STAT3特异性结合的小分子化合物芹黄素,然后运用Western-blot、双荧光素酶报告基因实验证实了芹黄素对STAT3的抑制作用。采用分子对接技术分析STAT3与芹黄素的结合位点,结果揭示芹黄素主要通过结合STAT3的SH2结构域抑制其磷酸化。本研究为芹黄素抗癌作用提供了理论基础,同时,也为发现STAT3及其他药物靶点的小分子抑制剂提供了研究经验。
-
表 1 L9(34)正交试验结果及直观分析(n = 3)
编号 A(%) B(w/V) C(t/h) D(T/ ℃) 对萼猕猴桃苷E含量(mg/g) 1 2 3 1 55(1) 1∶20(1) 0.5(1) 90(1) 7.94 7.98 7.87 2 55(1) 1∶25(2) 1(2) 95(2) 7.99 7.98 8.09 3 55(1) 1∶30(3) 1.5(3) 100(3) 7.89 7.85 7.98 4 65(2) 1∶20(1) 1(2) 100(3) 7.87 7.81 7.77 5 65(2) 1∶25(2) 1.5(3) 90(1) 7.85 7.99 7.97 6 65(2) 1∶30(3) 0.5(1) 95(2) 7.91 7.93 8.05 7 75(3) 1∶20(1) 1.5(3) 95(2) 7.91 7.91 7.81 8 75(3) 1∶25(2) 0.5(1) 100(3) 7.84 7.75 7.81 9 75(3) 1∶30(3) 1(2) 90(1) 7.88 7.97 7.87 K1 71.57 70.87 71.08 71.32 K2 71.15 71.27 71.23 71.58 K3 70.75 71.33 71.16 70.57 k1 7.95 7.87 7.90 7.92 k2 7.91 7.92 7.91 7.95 k3 7.86 7.93 7.91 7.84 R 0.09(2) 0.05(3) 0.02(4) 0.11(1)  下载: 导出CSV
下载: 导出CSV
表 2 对萼猕猴桃叶提取工艺方差分析
方差来源 离均差平方和 自由度 均方 F值 P值 A 0.037 2 0.019 4.989 0.019* B 0.014 2 0.007 1.856 0.185 C 0.001 2 0.001 0.167 0.847 D 0.061 2 0.031 8.161 0.003** 误差 0.067 18 0.004 合计 0.181 26 注:f1=2,f2=18,F0.05(2, 18)=3.55;*P<0.05,**P<0.01.
下载: 导出CSV
表 3 不同型号大孔树脂对对萼猕猴桃苷E静态吸附和解吸附的测定结果
树脂型号 吸附率(%) 解吸率(%) 权重(%) D101 91.54 96.67 88.49 AB-8 88.71 98.01 86.94 DM130 62.10 89.80 55.76 HPD100 87.82 97.25 85.41 NKA-9 87.15 97.64 85.09
下载: 导出CSV
-
[1] 中国植物志编委. 中国植物志[M]. 北京: 科学出版社, 1984: 218. [2] 国家中医药管理局《中华本草》编委会. 中华本草-1[M]. 上海: 上海科学技术出版社, 1999: 548-549. [3] 陈豪, 潘坤官, 何丽君. 抗癌中药猫人参研究概况[J]. 海峡药学, 2011, 23(12):9-11. doi: 10.3969/j.issn.1006-3765.2011.12.004 [4] 石垚, 贾敏, 张婷婷, 等. 不同生长时段对萼猕猴桃叶中黄酮类成分的含量分析[J]. 国际药学研究杂志, 2018, 45(10):795-798. [5] XIN H L, WU Y C, SU Y H, et al. Novel flavonoids from the leaves of Actinidia valvata Dunn: structural elucidation and antioxidant activity[J]. Planta Med, 2011, 77(1):70-73. doi: 10.1055/s-0030-1250113 [6] 辛海量. 抗心肌缺血的对萼猕猴桃叶组合物: CN201510213371.2[P]. 2018-08-21. [7] 辛海量, 凌昌全, 曲丽萍, 等. 对萼猕猴桃苷E、F在制备抗心肌缺血药物中的应用: CN201110271050. X[P]. 2013-02-06. [8] 李伟. 黄酮类化合物提取方法概述[J]. 农产品加工, 2023(14):75-78. [9] 陈丛瑾. 大孔吸附树脂分离纯化黄酮类化合物的研究进展[J]. 化学与生物工程, 2010, 27(11):1-4. doi: 10.3969/j.issn.1672-5425.2010.11.001 [10] 梁耀光, 吕巧莉. 葡聚糖凝胶Sephadex LH-20分离苷类的研究进展[J]. 广东化工, 2013, 40(13): 101, 93. 期刊类型引用(1)
1. 赵珊,张文青,张欣悦,阎星旭,钱文秀,李遇伯. 基于药物靶标识别中药活性成分的研究方法及应用. 药物评价研究. 2023(07): 1578-1586 .  百度学术
百度学术其他类型引用(2)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 1494
- HTML全文浏览量: 829
- PDF下载量: 2
- 被引次数: 3
