

 下载:
下载:
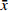
 下载:
下载:

-
西罗莫司(sirolimus,SRL),又称雷帕霉素,是第三代免疫抑制剂,在临床上常用于抑制肝、肾等器官移植后的免疫排斥反应。SRL属于生物药剂学分类Ⅱ类药物,在水中的溶解度极低,而渗透性良好[1-4]。SRL药理活性高,但因水溶性差,且易被肠壁和肝中的CYP3A4同工酶广泛代谢,致使其口服生物利用度较低。这是临床应用SRL的重要缺陷之一。目前,已上市的SRL制剂主要是纳米结晶片,生物利用度约为17%[5-7]。
通过适当的制剂技术提高SRL在胃肠道中的溶解度,可提高其口服生物利用度。在前期研究中,课题组分别独立进行了含SRL的自微乳(self-microemulsifying drug delivery system,SMEDDS)、固体分散体(solid dispersion,SD)和纳米结构脂质载体(nanostructured lipid carriers,NLC)的构建,均显著改善了SRL的体外溶出。本实验在前期研究的基础上,新增环糊精衍生物对SRL的增溶研究,结合体外溶出度和体内生物利用度,综合分析和评价各增溶制剂的优势和缺陷,从而为解决口服难溶性药物的研究提供参考。
-
Agilent 1200型高效液相色谱仪(美国Agilent公司);Starter 2C型pH计(上海奥豪斯仪器公司);RCZ-6BZ型药物溶出仪(上海黄海药检仪器公司);真空冷冻干燥箱(北京博医康试验仪器公司);NS1001L2K高压匀质机(意大利NiroSoavi公司);UV-2800AH型紫外可见分光光度仪(上海优尼科仪器有限公司);液相色谱-质谱联用仪(美国AB-SCIEX有限公司)。
-
SRL对照品(含量99.9%)、SRL原料药(含量99.6%),购自福建科瑞药业有限公司;子囊霉素对照品(上海齐奥化工科技有限公司),Rapamune®(美国惠氏制药)。聚乙二醇6000(PEG 6000)、聚乙烯吡咯烷酮(PVP K30)均购自国药集团化学试剂有限公司;聚氧乙烯-聚氧丙烯共聚物(Poloxamer 188)、聚氧乙烯35蓖麻油(Cremophor EL)、聚氧乙烯氢化蓖麻油(Cremophor RH40)均购自德国BASF公司;油酸聚乙二醇甘油酯(Labrafil M1944CS)、二乙二醇单乙基醚(Transcutol P)、辛酸癸酸聚乙二醇甘油酯(Labrasol)、棕榈酸硬脂酸甘油酯(Precirol ATO5)、月桂酸聚乙二醇甘油酯 (Gelucire 44/14)均购自法国GATTEFOSSE公司;HP-β-CD、DM-β-CD、SBE-β-CD(山东滨州智源生物科技有限公司)。
-
采用高效液相色谱仪(HPLC)测定样品中的SRL含量[8]。色谱柱为Eclipse XDB-C18(150 mm×4.6 mm,5 μm),流动相为乙腈-甲醇-水(45∶34∶21),流速为1 ml/min,检测波长为278 nm,柱温为50 ℃,进样量为20 μl。配制浓度为2、4、8、12、16、20 μg/ml的SRL对照品溶液,得标准曲线为Y=54.712X+1.221,r=0.999 9,表明在2~20 μg/ml浓度范围内线性关系良好。另外,精密度、回收率符合要求。
-
参考前期研究[9],称取1 g SRL原料药,加入19 g的助乳化剂Transcutol HP,超声至全部溶解后,加入22 g油相Labrafil M1944CS及39 g乳化剂Cremophor EL,涡旋混匀,得到淡黄色澄清溶液,即SRL-SMEDDS。
-
参考前期研究[10-11],取Gelucire44/14和Crodamol GTCC在75 ℃水浴中完全熔融后,加入SRL原料药搅拌均匀成澄明油相,再将同温度吐温−80的水溶液迅速倒入油相,以300 r/min搅拌30 min制备初乳,再经高压匀质机90 MPa乳匀5次,即得SRL-NLC分散液,其中SRL为0.21%,Gelucire44/14:Crodamol GTCC(1∶2.1),脂质总量为10%,吐温−80为7.33%。随后,将SRL-NLC(42.6%)加入微晶纤维素和聚乙烯吡咯烷酮(50%,4∶1)中,研磨混合并放置过夜以充分吸附,加入甘露醇(冻干保护剂,3%),经冷冻干燥过夜后,所得固体粉末中加入低取代羟丙基纤维素(崩解剂,4%)和二氧化硅(助流剂,0.4%)即得固化纳米脂质体。
-
采用溶剂-熔融法制备SRL-SD。称取载体材料,于80 ℃水浴加热熔融,滴入SRL乙醇溶液,充分混匀,待乙醇挥发完全后,迅速将其倾倒于冰浴条件下的不锈钢板上成薄膜,固化,再于−18 ℃放置4 h后,将固体分散体从不锈钢板上刮下,置真空干燥器中干燥,待脆化后研细,过80目筛,即得SRL-SD。以载体种类、药物-载体比例为考察因素,以0.4% SDS中的溶出度为指标,对SRL-SD进行单因素分析。
-
称取适量β-环糊精衍生物溶于去离子水中,缓慢滴加SRL乙醇溶液,在一定温度下磁力搅拌至澄清透明,减压挥发4 h,使乙醇挥发完全,再置于4 ℃冰箱冷藏12 h,降低SRL的溶解度,从而使游离的SRL发生结晶。经0.22 μm微孔滤膜过滤除去结晶,滤液冷冻干燥24 h,所得固体研磨细化,过80目筛,即得SRL-IC。
称取一定量的SRL-IC置10 ml容量瓶中,加入50%甲醇水溶液,超声至全部溶解后,定容至刻度,并采用HPLC测定SRL含量,根据公式:包封率(%)=[(SRL投入量-SRL测定量)/ SRL投入量]×100%,进行计算。以环糊精衍生物的种类、浓度、温度、乙醇体积和投药量为考察因素,以包封率为指标,对SRL-IC进行单因素分析。
-
参考《中国药典》2015年版四部通则0931项下溶出度与释放度测定法,考察SRL原料药、市售片(Rapamune®)、SRL-SMEDDS、SRL-NLC、SRL-IC及SRL-SD的溶出曲线。除市售片外,其余样品均装入硬胶囊中,每个胶囊含1 mg SRL。采用桨法,搅拌速度为100 r/min,溶出介质体积为250 ml,分别以0.4% SDS、水、pH 1.2盐酸溶液、pH 4.5醋酸盐缓冲液、pH 6.8磷酸盐缓冲液、pH 7.4磷酸盐缓冲液为溶出介质。将两颗胶囊或药片置于沉降篮中,投入溶出介质,在10、30、45、60、90、120 min,吸取2 ml介质,并补充等温等体积的介质,采用HPLC测定样品中的药物含量,绘制溶出曲线。
-
选用比格犬为实验动物,采用6周期6交叉实验设计,进行SRL原料药、市售片(Rapamune®)、SRL-SMEDDS、SRL-NLC、SRL-IC及SRL-SD的药代动力学试验。给药剂量为1 mg SRL,实验动物试验开始前12 h禁食不禁水,给药4 h后自由饮水,2次给药间隔2周以上的清洗期。于给药前,0.25、0.5、0.75、1、1.5、2、3、4、6、8、10、12、24、36、48及72 h分别经前肢小静脉采血2 ml,置于含肝素和EDTA的抗凝管中,−20 ℃保存备用。血样处理与测定方法参照课题组前期研究[12]。
-
如图1A所示,不同载体材料制备的SRL-SD的溶出曲线显示了明显的差异,溶出速率为PEG6000>F68>PVP K30>HPMC606>HPMC-AS-MF。同时,各载体材料的溶出度均不理想(≤50%),因此进一步考察采用二元载体制备SRL-SD。
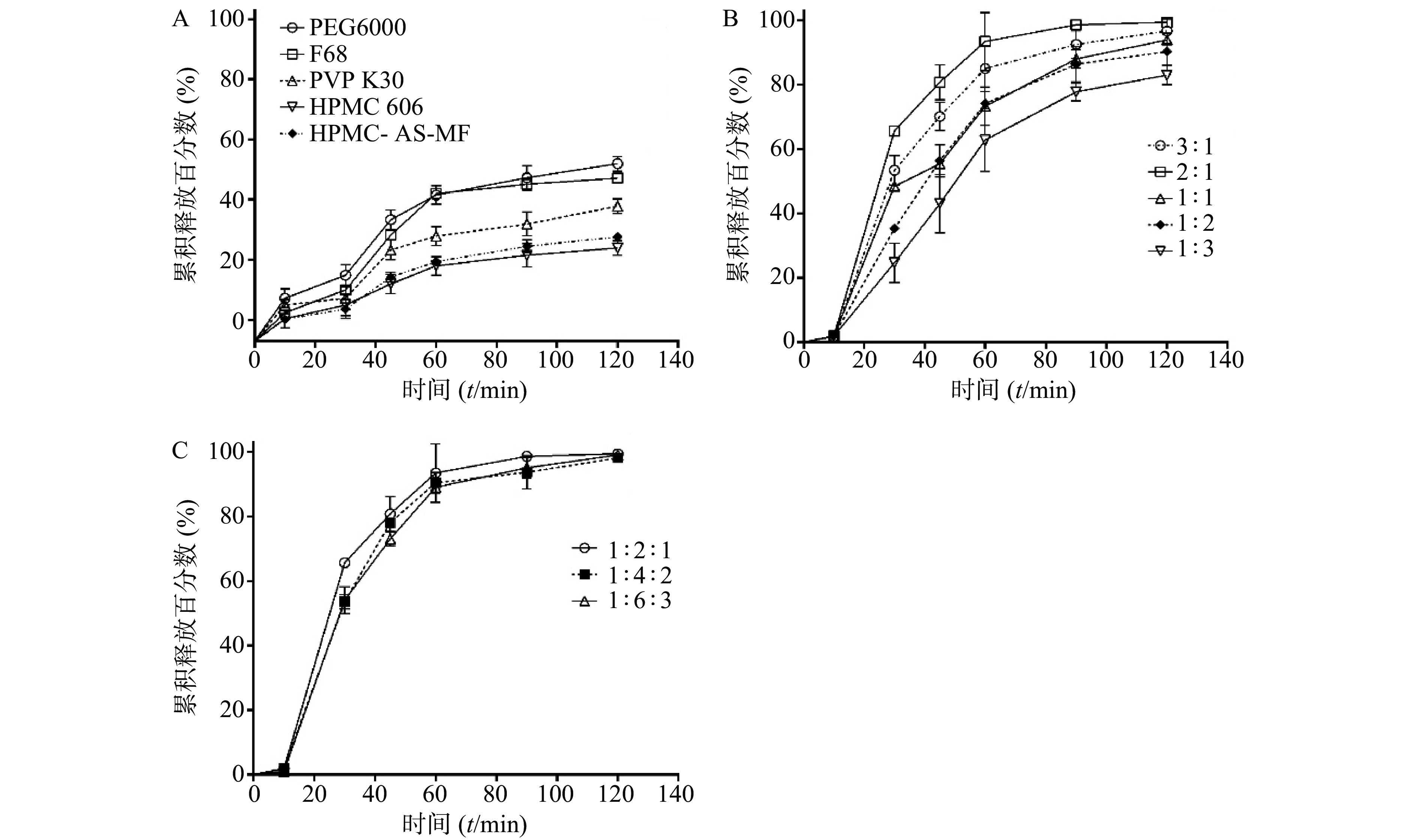
图 1 单因素考察固体分散体的制备对体外溶出曲线的影响
选择PEG6000联合F68制备二元载体固体分散体[13],两者比例为3∶1、2∶1、1∶1、1∶2、1∶3。随PEG6000/F68比例的增大,则SRL溶出度呈增大趋势,在PEG6000/F68为2∶1时的溶出度达到最大(图1B)。
-
在PEG6000/F68=2∶1的基础上,进一步考察药物-载体比例对SRL-SD溶出的影响。药物-PEG6000/F68载体比例为1∶2∶1、1∶4∶2及1∶6∶3所制的SRL-SD的溶出曲线相似,没有明显差别,2 h的溶出度都接近100%(图1C)。因此优选载药量最大,即药物- PEG6000/F68载体比例为1∶2∶1。
-
在其他条件相同的情况下,HP-β-CD、SBE-β-CD和DM-β-CD对SRL的包封率分别为(11.21±3.35)%、(8.24±3.11)%和(31.86±3.26)%,见图2A。因此,优选DM-β-CD制备SRL-IC。
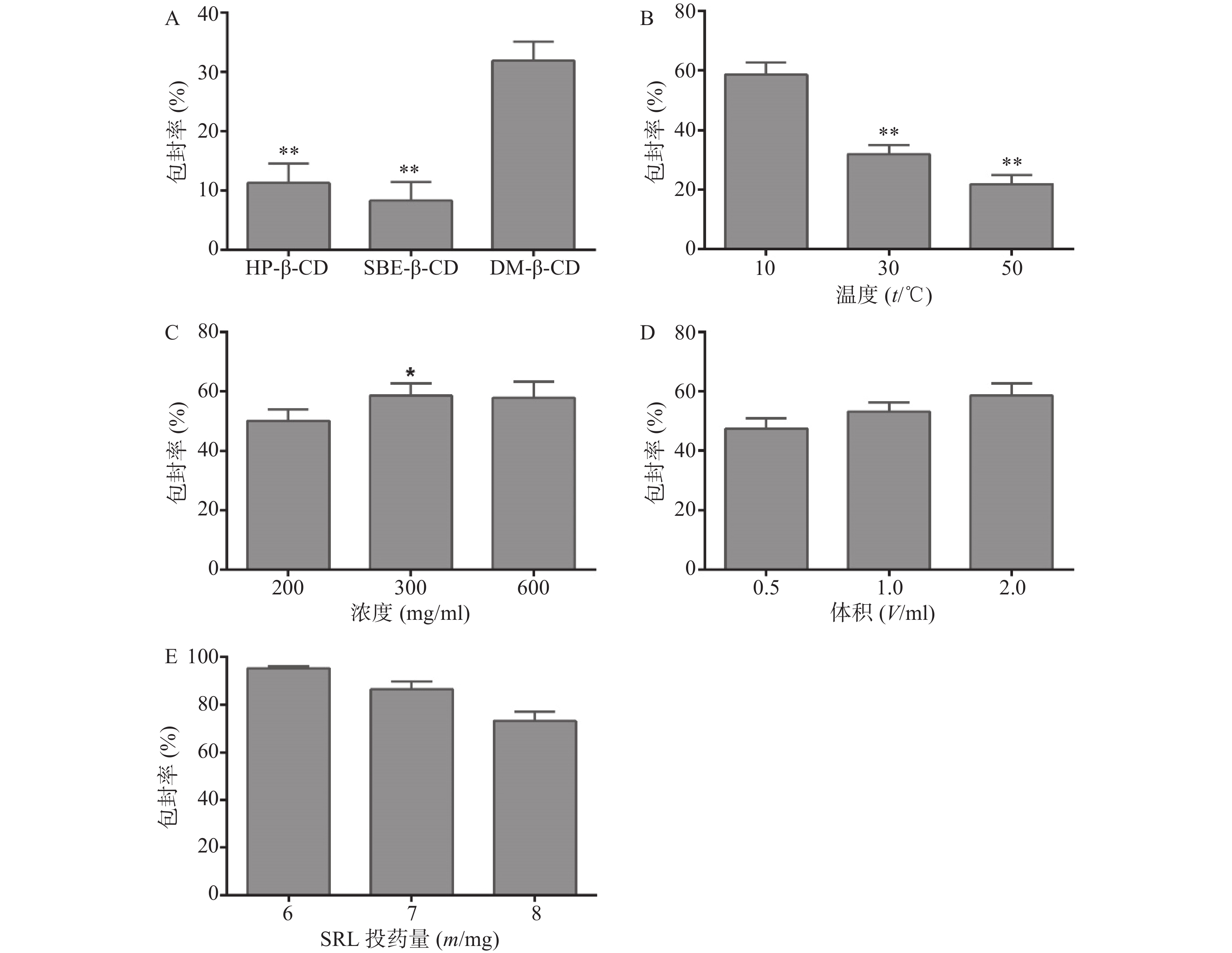
图 2 单因素考察包合物的制备工艺对包合率的影响
-
采用DM-β-CD制备SRL-IC,考察不同温度对包封率的影响。结果显示(图2B),温度越低,包封率越高,10 ℃条件下制备的SRL-IC的包封率显著高于30 ℃和50 ℃(P<0.01),为(58.61±4.16)%。因此,优选10 ℃制备SRL-IC。
-
DM-β-CD的浓度由200 mg/ml增大至300 mg/ml,SRL的包封率由(52.12±4.17)%增大至(58.61±4.11)%(P<0.05,图2C)。进一步增大DM-β-CD的浓度至600 mg/ml,包封率没有明显变化(P>0.05)。因此,优选DM-β-CD的浓度为300 mg/ml制备SRL-IC。
-
乙醇体积由0.5 ml增大至2 ml,包封率呈增大趋势(图2D)。因此,优选乙醇体积为0.5 ml制备SRL-IC。
-
SRL的投药量6 mg增大至8 mg,包封率显著降低,6 mg SRL的包封率为(95.21±1.10)%,见图2E。因此,优选SRL的投药量为6 mg。
-
考察SRL-SD、SRL-IC、SRL-SMEDDS及SRL-NLC在不同介质中的溶出曲线。如图3所示,在0.4% SDS中,各制剂在2 h的溶出度均超过80%,尤其是SMEDDS和NLC的溶出度接近100%。
图 3 不同增溶制剂的西罗莫司在溶出介质中的溶出曲线图(n=3)
在pH 6.8和水中,SRL-SD的溶出速率减小,2 h的溶出度分别为(65.00±4.90)%和(76.70±1.95)%。在pH 4.5和pH 7.4的介质中,SRL-SD的溶出在1 h达到最大值,分别为(53.20±4.34)%和(55.20±4.34)%,随后溶出度逐渐降低。在pH 1.2的介质中,未检测到SRL。
在水、pH 4.5、pH 6.8和pH 7.4中,SRL-IC在40 min内的溶出速率有所减小,但2 h的累积溶出没有明显变化,均在80%以上。在pH 1.2的介质中,SRL-IC的溶出度在30 min达到最大值,为(49.84±7.21)%,随后溶出度逐渐降低。
SRL-SMEDDS和SRL-NLC显示了与SRL-SD相似的溶出趋势,即在水和pH 6.8中的溶出度低于0.4% SDS,但大于80%。在pH 4.5和pH 7.4的介质中,溶出达到峰值(约80%)后逐渐降低。
-
SRL血药浓度-时间曲线见图4,经DAS 3.2.6软件处理后,具体参数见表 1。
表 1 非房室模型体内药动学参数(
$ \bar x$ ±s)参数 SRL SRL-SD SRL-IC SRL- NLC SRL-SMEDDS Rapamune® AUC0→72(µg·h/ml) 0.70±0.13 2.06±0.79 3.66±2.64 8.60±2.03 10.76±1.57 11.02±2.73 AUC0→t(µg·h/ml) 0.73±0.15 2.07±0.81 3.78±2.84 8.67±1.95 11.15±2.11 11.75±3.13 t1/2 (t/h) 16.53±1.50 14.50±2.15 20.64±5.45 8.97±6.87 12.97±5.67 14.54±5.67 tmax(t/h) 1.04±0.25 1.25±0.28 1.04±0.25 1.13±0.31 1.50±0.38 1.83±0.26 cmax (ng/ml) 0.16±0.05 0.36±0.05 0.53±0.13 0.90±0.09 1.23±0.07 1.28±0.13 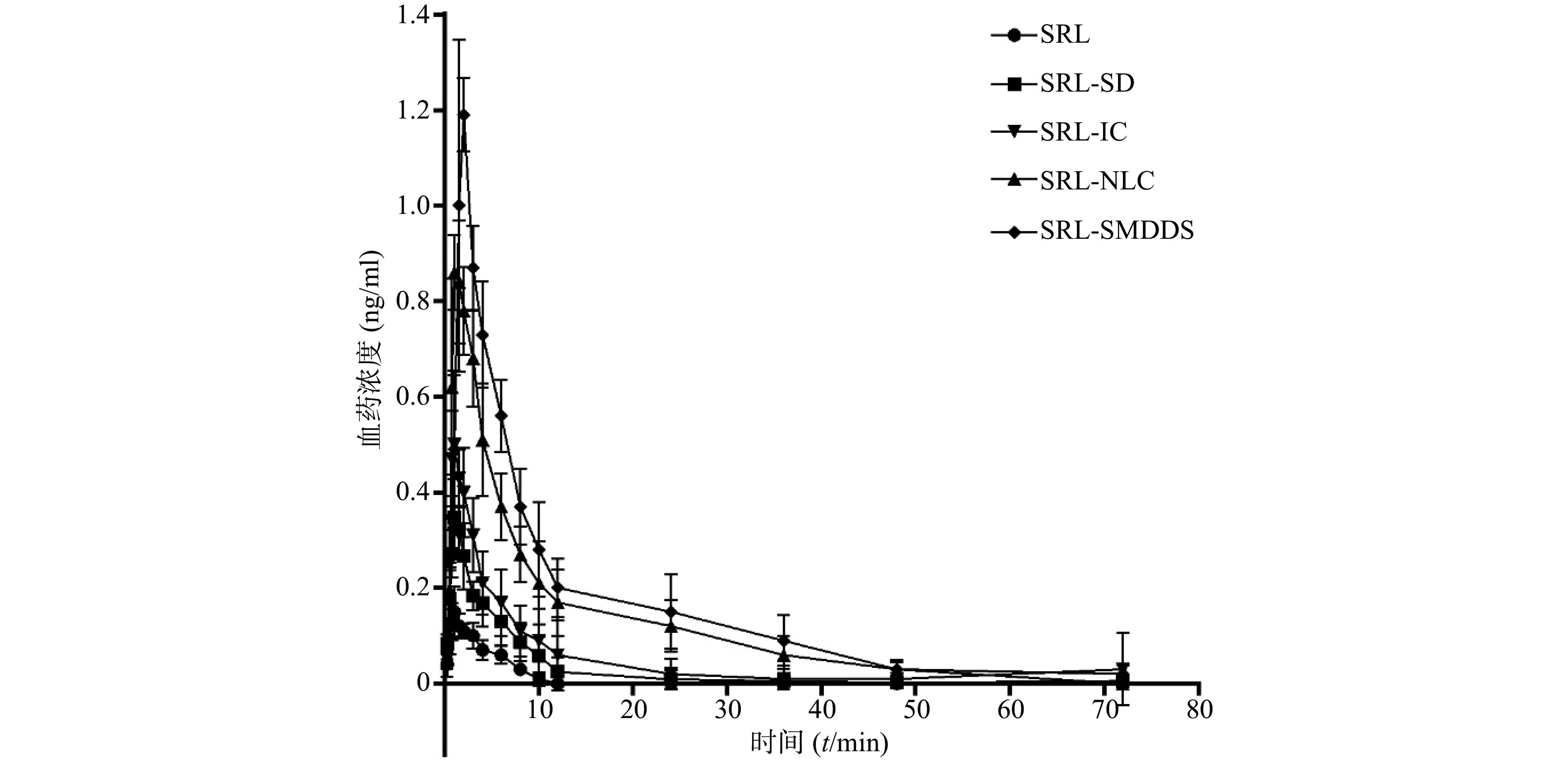
图 4 参比制剂与受试试剂平均血药浓度-时间曲线 (n=6)
以原料药为参比制剂,SRL-SD、SRL-IC、SRL-SMEDDS、SRL-NLC、Rapamune®的相对生物利用度分别为332.8%、522.9%、1 228.6%、1 537.1%、1 574.3%,表明各增溶方法都显著提高了SRL的生物利用度。
以市售纳米晶片Rapamune®为参比制剂,SRL-SD、SRL-IC、SRL-NLC、SRL-SMEDDS的相对生物利用度分别为18.7%、33.2%、78.0%、97.6%,可见在各增溶方法中,SMEDDS对SRL体内吸收的作用最显著,与市售制剂相当。
-
本研究同时制备和比较了SRL的4种增溶制剂,均显示了良好的体外溶出度。同时,各制剂都提高了SRL的生物利用度,但体内吸收程度有较明显的差异。
首先,SRL本身的性质是影响体内吸收的重要因素。在理化性质方面,SRL在电解质溶液中可发生开环水解,特别是在强酸和碱性条件下,降解速率显著增加[14]。在生理因素方面,SRL是肠道内CYP3A4酶和P糖蛋白的底物,对肠道吸收有较大影响[15]。
其次,制剂本身的特点对体内吸收有重要影响。SMEDDS和NLC均可形成纳米级的脂质微粒,在胃肠道消化后可形成乳糜胶束[16-17]均减轻了胃肠液的pH对SRL的降解作用,因此SMEDDS和NLC对脂质微粒中的SRL有一定的保护作用。相比之下,SD中的SRL快速释放后,载体材料失去了对药物的隔离保护作用,导致SRL在极短的时间内发生降解。另外,环糊精的空腔可以容纳药物分子[18],不仅提高了SRL的溶解度,而且降低了H+和OH-对SRL的作用概率,减缓了SRL的降解。本研究的体外溶出试验也证实了不同增溶制剂中SRL稳定性的差异。
同时,SMEDDS的辅料Labrafil M1944 CS和Cremophor EL[9, 19-21]和NLC中的脂质及其代谢产物能够抑制CYP3A4酶的代谢和P糖蛋白外排,消化后形成的乳糜胶束还可通过淋巴途径吸收[22],从而提高了生物利用度[10-11]。
另外,由于SRL分子量较大,分子结构可能仅有部分插入环糊精的空腔中。因此,尽管环糊精提高了SRL的溶出度,但包合物的稳定性较差,进入胃肠道后,药物可被胃肠液中的成分替换[23],导致SRL加速降解或发生重结晶,进而生物利用度下降。
Study on sirolimus solubilization technology based on in vitro dissolution and in vivo bioavailability
-
摘要:
目的 评价不同增溶技术对西罗莫司(sirolimus,SRL)的体外溶出与体内吸收的影响。 方法 选取固体分散体(SD)、包合物(IC)、自微乳(SMEDDS)和纳米结构脂质载体(NLC)为SRL的增溶技术。SRL-SMEDDS和SRL-NLC已在前期研究中获得最优处方。另外,以包封率、体外溶出度等为指标,筛选SRL-SD和SRL-IC的处方工艺。分别采用0.4% SDS,水,及pH 1.2、pH 4.5、pH 6.8、pH 7.4缓冲液为溶出介质,考察市售制剂Rapamune®,以及自制的各增溶制剂的溶出曲线。采用比格犬体内药动学试验,考察上述制剂的体内吸收度。 结果 在0.4% 十二烷基硫酸钠(SDS)中,各制剂在2 h的溶出度均超过80%。在pH 1.2的介质中,无法测得SRL-SD的溶出度,而IC、SMEDDS和NLC的溶出度呈先增大后减小的趋势。在其他介质中,SRL的溶出度均有所降低,而SRL-IC显示了最佳的溶出度,未出现明显的降低趋势。体内药动学试验结果显示,原料药、SRL-SD、SRL-IC、SRL-NLC和SRL-SMEDDS的相对生物利用度分别为9.1%、18.7%、33.2%、78.0%、97.6%。 结论 SD、SMEDDS、NLC、IC均可提高SRL的体外溶出度和体内吸收度,其中,SMEDDS对SRL的生物利用度改善最为明显。 Abstract:Objective To evaluate the effects of different solubilizing techniques on the in vitro dissolution and in vivo pharmacokinetics of Sirolimus (SRL). Methods Solid dispersions (SD), inclusion complex (IC), self-micro emulsifying drug delivery system (SMEDDS) and nano-structured lipid carrier (NLC) were selected as the solubilization technology for SRL. SRL-SMEDDS and SRL-NLC have obtained the optimal prescription in the previous studies. Additionally, the formulation process of SRL-SD and SRL-IC was screened by using inclusion rate and dissolution profiles as indicators. 0.4% SDS, water and buffer solutions with pH 1.2, 4.5, 6.8, 7.4 were used as dissolution media. The dissolution profile of the commercially available formulation Rapamune® and the lab-made solubilized preparations were investigated. The in vivo absorption of the above preparations was examined using a pharmacokinetic test in Beagle dogs. Results In 0.4% SDS, the dissolution of each preparation exceeded 80% in 2 h. In the medium of pH 1.2, the dissolution of SRL-SD could not be measured while the dissolution of IC, SMEDDS and NLC increased first and then decreased. In other media, the dissolution of the SRL was reduced. The SRL-IC showed the best dissolution without a significant decrease. The relative bioavailability of APIs, SRL-SD, SRL-IC, SRL-NLC and SRL-SMEDDS were 9.1%, 18.7%, 33.2%, 78.0%, and 97.6% respectively in vivo pharmacokinetic tests. Conclusion SD, SMEDDS, NLC, and IC can improve the in vitro dissolution and in vivo absorption of SRL. Among them, SMEDDS has the most significant improvement in the bioavailability of SRL. -
Key words:
- sirolimus /
- solid dispersion /
- inclusion complex /
- SMEDDS /
- nano-structured lipid carrier /
- dissolution /
- bioavailability
-
西罗莫司(sirolimus,SRL),又称雷帕霉素,是第三代免疫抑制剂,在临床上常用于抑制肝、肾等器官移植后的免疫排斥反应。SRL属于生物药剂学分类Ⅱ类药物,在水中的溶解度极低,而渗透性良好[1-4]。SRL药理活性高,但因水溶性差,且易被肠壁和肝中的CYP3A4同工酶广泛代谢,致使其口服生物利用度较低。这是临床应用SRL的重要缺陷之一。目前,已上市的SRL制剂主要是纳米结晶片,生物利用度约为17%[5-7]。
通过适当的制剂技术提高SRL在胃肠道中的溶解度,可提高其口服生物利用度。在前期研究中,课题组分别独立进行了含SRL的自微乳(self-microemulsifying drug delivery system,SMEDDS)、固体分散体(solid dispersion,SD)和纳米结构脂质载体(nanostructured lipid carriers,NLC)的构建,均显著改善了SRL的体外溶出。本实验在前期研究的基础上,新增环糊精衍生物对SRL的增溶研究,结合体外溶出度和体内生物利用度,综合分析和评价各增溶制剂的优势和缺陷,从而为解决口服难溶性药物的研究提供参考。
1. 仪器与试剂
1.1 仪器
Agilent 1200型高效液相色谱仪(美国Agilent公司);Starter 2C型pH计(上海奥豪斯仪器公司);RCZ-6BZ型药物溶出仪(上海黄海药检仪器公司);真空冷冻干燥箱(北京博医康试验仪器公司);NS1001L2K高压匀质机(意大利NiroSoavi公司);UV-2800AH型紫外可见分光光度仪(上海优尼科仪器有限公司);液相色谱-质谱联用仪(美国AB-SCIEX有限公司)。
1.2 试剂
SRL对照品(含量99.9%)、SRL原料药(含量99.6%),购自福建科瑞药业有限公司;子囊霉素对照品(上海齐奥化工科技有限公司),Rapamune®(美国惠氏制药)。聚乙二醇6000(PEG 6000)、聚乙烯吡咯烷酮(PVP K30)均购自国药集团化学试剂有限公司;聚氧乙烯-聚氧丙烯共聚物(Poloxamer 188)、聚氧乙烯35蓖麻油(Cremophor EL)、聚氧乙烯氢化蓖麻油(Cremophor RH40)均购自德国BASF公司;油酸聚乙二醇甘油酯(Labrafil M1944CS)、二乙二醇单乙基醚(Transcutol P)、辛酸癸酸聚乙二醇甘油酯(Labrasol)、棕榈酸硬脂酸甘油酯(Precirol ATO5)、月桂酸聚乙二醇甘油酯 (Gelucire 44/14)均购自法国GATTEFOSSE公司;HP-β-CD、DM-β-CD、SBE-β-CD(山东滨州智源生物科技有限公司)。
2. 方法
2.1 SRL含量测定方法
采用高效液相色谱仪(HPLC)测定样品中的SRL含量[8]。色谱柱为Eclipse XDB-C18(150 mm×4.6 mm,5 μm),流动相为乙腈-甲醇-水(45∶34∶21),流速为1 ml/min,检测波长为278 nm,柱温为50 ℃,进样量为20 μl。配制浓度为2、4、8、12、16、20 μg/ml的SRL对照品溶液,得标准曲线为Y=54.712X+1.221,r=0.999 9,表明在2~20 μg/ml浓度范围内线性关系良好。另外,精密度、回收率符合要求。
2.2 SRL增溶方法
2.2.1 SRL-SMEDDS的制备
参考前期研究[9],称取1 g SRL原料药,加入19 g的助乳化剂Transcutol HP,超声至全部溶解后,加入22 g油相Labrafil M1944CS及39 g乳化剂Cremophor EL,涡旋混匀,得到淡黄色澄清溶液,即SRL-SMEDDS。
2.2.2 SRL-NLC的制备
参考前期研究[10-11],取Gelucire44/14和Crodamol GTCC在75 ℃水浴中完全熔融后,加入SRL原料药搅拌均匀成澄明油相,再将同温度吐温−80的水溶液迅速倒入油相,以300 r/min搅拌30 min制备初乳,再经高压匀质机90 MPa乳匀5次,即得SRL-NLC分散液,其中SRL为0.21%,Gelucire44/14:Crodamol GTCC(1∶2.1),脂质总量为10%,吐温−80为7.33%。随后,将SRL-NLC(42.6%)加入微晶纤维素和聚乙烯吡咯烷酮(50%,4∶1)中,研磨混合并放置过夜以充分吸附,加入甘露醇(冻干保护剂,3%),经冷冻干燥过夜后,所得固体粉末中加入低取代羟丙基纤维素(崩解剂,4%)和二氧化硅(助流剂,0.4%)即得固化纳米脂质体。
2.2.3 SRL-SD的制备
采用溶剂-熔融法制备SRL-SD。称取载体材料,于80 ℃水浴加热熔融,滴入SRL乙醇溶液,充分混匀,待乙醇挥发完全后,迅速将其倾倒于冰浴条件下的不锈钢板上成薄膜,固化,再于−18 ℃放置4 h后,将固体分散体从不锈钢板上刮下,置真空干燥器中干燥,待脆化后研细,过80目筛,即得SRL-SD。以载体种类、药物-载体比例为考察因素,以0.4% SDS中的溶出度为指标,对SRL-SD进行单因素分析。
2.2.4 SRL-IC的制备
称取适量β-环糊精衍生物溶于去离子水中,缓慢滴加SRL乙醇溶液,在一定温度下磁力搅拌至澄清透明,减压挥发4 h,使乙醇挥发完全,再置于4 ℃冰箱冷藏12 h,降低SRL的溶解度,从而使游离的SRL发生结晶。经0.22 μm微孔滤膜过滤除去结晶,滤液冷冻干燥24 h,所得固体研磨细化,过80目筛,即得SRL-IC。
称取一定量的SRL-IC置10 ml容量瓶中,加入50%甲醇水溶液,超声至全部溶解后,定容至刻度,并采用HPLC测定SRL含量,根据公式:包封率(%)=[(SRL投入量-SRL测定量)/ SRL投入量]×100%,进行计算。以环糊精衍生物的种类、浓度、温度、乙醇体积和投药量为考察因素,以包封率为指标,对SRL-IC进行单因素分析。
2.3 体外溶出试验
参考《中国药典》2015年版四部通则0931项下溶出度与释放度测定法,考察SRL原料药、市售片(Rapamune®)、SRL-SMEDDS、SRL-NLC、SRL-IC及SRL-SD的溶出曲线。除市售片外,其余样品均装入硬胶囊中,每个胶囊含1 mg SRL。采用桨法,搅拌速度为100 r/min,溶出介质体积为250 ml,分别以0.4% SDS、水、pH 1.2盐酸溶液、pH 4.5醋酸盐缓冲液、pH 6.8磷酸盐缓冲液、pH 7.4磷酸盐缓冲液为溶出介质。将两颗胶囊或药片置于沉降篮中,投入溶出介质,在10、30、45、60、90、120 min,吸取2 ml介质,并补充等温等体积的介质,采用HPLC测定样品中的药物含量,绘制溶出曲线。
2.4 体内药代动力学试验
选用比格犬为实验动物,采用6周期6交叉实验设计,进行SRL原料药、市售片(Rapamune®)、SRL-SMEDDS、SRL-NLC、SRL-IC及SRL-SD的药代动力学试验。给药剂量为1 mg SRL,实验动物试验开始前12 h禁食不禁水,给药4 h后自由饮水,2次给药间隔2周以上的清洗期。于给药前,0.25、0.5、0.75、1、1.5、2、3、4、6、8、10、12、24、36、48及72 h分别经前肢小静脉采血2 ml,置于含肝素和EDTA的抗凝管中,−20 ℃保存备用。血样处理与测定方法参照课题组前期研究[12]。
3. 结果
3.1 SRL-SD的制备
3.1.1 载体种类
如图1A所示,不同载体材料制备的SRL-SD的溶出曲线显示了明显的差异,溶出速率为PEG6000>F68>PVP K30>HPMC606>HPMC-AS-MF。同时,各载体材料的溶出度均不理想(≤50%),因此进一步考察采用二元载体制备SRL-SD。
选择PEG6000联合F68制备二元载体固体分散体[13],两者比例为3∶1、2∶1、1∶1、1∶2、1∶3。随PEG6000/F68比例的增大,则SRL溶出度呈增大趋势,在PEG6000/F68为2∶1时的溶出度达到最大(图1B)。
3.1.2 药物-载体比例
在PEG6000/F68=2∶1的基础上,进一步考察药物-载体比例对SRL-SD溶出的影响。药物-PEG6000/F68载体比例为1∶2∶1、1∶4∶2及1∶6∶3所制的SRL-SD的溶出曲线相似,没有明显差别,2 h的溶出度都接近100%(图1C)。因此优选载药量最大,即药物- PEG6000/F68载体比例为1∶2∶1。
3.2 SRL-IC的制备
3.2.1 β-环糊精衍生物种类
在其他条件相同的情况下,HP-β-CD、SBE-β-CD和DM-β-CD对SRL的包封率分别为(11.21±3.35)%、(8.24±3.11)%和(31.86±3.26)%,见图2A。因此,优选DM-β-CD制备SRL-IC。
图 2 单因素考察包合物的制备工艺对包合率的影响A.β-环糊精衍生物种类的影响,**P<0.01,与DM-β-CD比较;B. 温度的影响,**P<0.01,与10 ℃比较;C. 环糊精衍生物浓度的影响,*P<0.05,与200 mg/ml比较;D. 乙醇体积的影响;E. SRL投药量的影响3.2.2 温度
采用DM-β-CD制备SRL-IC,考察不同温度对包封率的影响。结果显示(图2B),温度越低,包封率越高,10 ℃条件下制备的SRL-IC的包封率显著高于30 ℃和50 ℃(P<0.01),为(58.61±4.16)%。因此,优选10 ℃制备SRL-IC。
3.2.3 环糊精衍生物浓度
DM-β-CD的浓度由200 mg/ml增大至300 mg/ml,SRL的包封率由(52.12±4.17)%增大至(58.61±4.11)%(P<0.05,图2C)。进一步增大DM-β-CD的浓度至600 mg/ml,包封率没有明显变化(P>0.05)。因此,优选DM-β-CD的浓度为300 mg/ml制备SRL-IC。
3.2.4 乙醇体积
乙醇体积由0.5 ml增大至2 ml,包封率呈增大趋势(图2D)。因此,优选乙醇体积为0.5 ml制备SRL-IC。
3.2.5 投药量
SRL的投药量6 mg增大至8 mg,包封率显著降低,6 mg SRL的包封率为(95.21±1.10)%,见图2E。因此,优选SRL的投药量为6 mg。
3.3 体外溶出度
考察SRL-SD、SRL-IC、SRL-SMEDDS及SRL-NLC在不同介质中的溶出曲线。如图3所示,在0.4% SDS中,各制剂在2 h的溶出度均超过80%,尤其是SMEDDS和NLC的溶出度接近100%。
在pH 6.8和水中,SRL-SD的溶出速率减小,2 h的溶出度分别为(65.00±4.90)%和(76.70±1.95)%。在pH 4.5和pH 7.4的介质中,SRL-SD的溶出在1 h达到最大值,分别为(53.20±4.34)%和(55.20±4.34)%,随后溶出度逐渐降低。在pH 1.2的介质中,未检测到SRL。
在水、pH 4.5、pH 6.8和pH 7.4中,SRL-IC在40 min内的溶出速率有所减小,但2 h的累积溶出没有明显变化,均在80%以上。在pH 1.2的介质中,SRL-IC的溶出度在30 min达到最大值,为(49.84±7.21)%,随后溶出度逐渐降低。
SRL-SMEDDS和SRL-NLC显示了与SRL-SD相似的溶出趋势,即在水和pH 6.8中的溶出度低于0.4% SDS,但大于80%。在pH 4.5和pH 7.4的介质中,溶出达到峰值(约80%)后逐渐降低。
3.4 比格犬体内药动学试验
SRL血药浓度-时间曲线见图4,经DAS 3.2.6软件处理后,具体参数见表 1。
表 1 非房室模型体内药动学参数($ \bar x$ ±s)参数 SRL SRL-SD SRL-IC SRL- NLC SRL-SMEDDS Rapamune® AUC0→72(µg·h/ml) 0.70±0.13 2.06±0.79 3.66±2.64 8.60±2.03 10.76±1.57 11.02±2.73 AUC0→t(µg·h/ml) 0.73±0.15 2.07±0.81 3.78±2.84 8.67±1.95 11.15±2.11 11.75±3.13 t1/2 (t/h) 16.53±1.50 14.50±2.15 20.64±5.45 8.97±6.87 12.97±5.67 14.54±5.67 tmax(t/h) 1.04±0.25 1.25±0.28 1.04±0.25 1.13±0.31 1.50±0.38 1.83±0.26 cmax (ng/ml) 0.16±0.05 0.36±0.05 0.53±0.13 0.90±0.09 1.23±0.07 1.28±0.13 以原料药为参比制剂,SRL-SD、SRL-IC、SRL-SMEDDS、SRL-NLC、Rapamune®的相对生物利用度分别为332.8%、522.9%、1 228.6%、1 537.1%、1 574.3%,表明各增溶方法都显著提高了SRL的生物利用度。
以市售纳米晶片Rapamune®为参比制剂,SRL-SD、SRL-IC、SRL-NLC、SRL-SMEDDS的相对生物利用度分别为18.7%、33.2%、78.0%、97.6%,可见在各增溶方法中,SMEDDS对SRL体内吸收的作用最显著,与市售制剂相当。
4. 讨论
本研究同时制备和比较了SRL的4种增溶制剂,均显示了良好的体外溶出度。同时,各制剂都提高了SRL的生物利用度,但体内吸收程度有较明显的差异。
首先,SRL本身的性质是影响体内吸收的重要因素。在理化性质方面,SRL在电解质溶液中可发生开环水解,特别是在强酸和碱性条件下,降解速率显著增加[14]。在生理因素方面,SRL是肠道内CYP3A4酶和P糖蛋白的底物,对肠道吸收有较大影响[15]。
其次,制剂本身的特点对体内吸收有重要影响。SMEDDS和NLC均可形成纳米级的脂质微粒,在胃肠道消化后可形成乳糜胶束[16-17]均减轻了胃肠液的pH对SRL的降解作用,因此SMEDDS和NLC对脂质微粒中的SRL有一定的保护作用。相比之下,SD中的SRL快速释放后,载体材料失去了对药物的隔离保护作用,导致SRL在极短的时间内发生降解。另外,环糊精的空腔可以容纳药物分子[18],不仅提高了SRL的溶解度,而且降低了H+和OH-对SRL的作用概率,减缓了SRL的降解。本研究的体外溶出试验也证实了不同增溶制剂中SRL稳定性的差异。
同时,SMEDDS的辅料Labrafil M1944 CS和Cremophor EL[9, 19-21]和NLC中的脂质及其代谢产物能够抑制CYP3A4酶的代谢和P糖蛋白外排,消化后形成的乳糜胶束还可通过淋巴途径吸收[22],从而提高了生物利用度[10-11]。
另外,由于SRL分子量较大,分子结构可能仅有部分插入环糊精的空腔中。因此,尽管环糊精提高了SRL的溶出度,但包合物的稳定性较差,进入胃肠道后,药物可被胃肠液中的成分替换[23],导致SRL加速降解或发生重结晶,进而生物利用度下降。
-
图 2 单因素考察包合物的制备工艺对包合率的影响
A.β-环糊精衍生物种类的影响,**P<0.01,与DM-β-CD比较;B. 温度的影响,**P<0.01,与10 ℃比较;C. 环糊精衍生物浓度的影响,*P<0.05,与200 mg/ml比较;D. 乙醇体积的影响;E. SRL投药量的影响
表 1 非房室模型体内药动学参数(
$ \bar x$ ±s)参数 SRL SRL-SD SRL-IC SRL- NLC SRL-SMEDDS Rapamune® AUC0→72(µg·h/ml) 0.70±0.13 2.06±0.79 3.66±2.64 8.60±2.03 10.76±1.57 11.02±2.73 AUC0→t(µg·h/ml) 0.73±0.15 2.07±0.81 3.78±2.84 8.67±1.95 11.15±2.11 11.75±3.13 t1/2 (t/h) 16.53±1.50 14.50±2.15 20.64±5.45 8.97±6.87 12.97±5.67 14.54±5.67 tmax(t/h) 1.04±0.25 1.25±0.28 1.04±0.25 1.13±0.31 1.50±0.38 1.83±0.26 cmax (ng/ml) 0.16±0.05 0.36±0.05 0.53±0.13 0.90±0.09 1.23±0.07 1.28±0.13  下载: 导出CSV
下载: 导出CSV
-
[1] RIAL M D E L C, ABBUD-FILHO M, GONÇALVES R T, et al. Individualizing early use of sirolimus in renal transplantation[J]. Transplant Proc,2010,42(10):4518-4525. doi: 10.1016/j.transproceed.2010.10.015 [2] TOSO C, MERANI S, BIGAM D L, et al. Sirolimus-based immunosuppression is associated with increased survival after liver transplantation for hepatocellular carcinoma[J]. Hepatology,2010,51(4):1237-1243. doi: 10.1002/hep.23437 [3] CHATEL M A, LARKIN D F. Sirolimus and mycophenolate as combination prophylaxis in corneal transplant recipients at high rejection risk[J]. Am J Ophthalmol,2010,150(2):179-184. doi: 10.1016/j.ajo.2010.03.010 [4] POUTON C W. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system[J]. Eur J Pharm Sci,2006,29(3-4):278-287. doi: 10.1016/j.ejps.2006.04.016 [5] RICCIUTELLI M, DI MARTINO P, BARBONI L, et al. Evaluation of rapamycin chemical stability in volatile-organic solvents by HPLC[J]. J Pharm Biomed Anal,2006,41(3):1070-1074. doi: 10.1016/j.jpba.2006.02.009 [6] BALAKRISHNAN P, LEE B, OH D H, et al. Enhanced oral bioavailability of dexibuprofen by a novel solid Self-emulsifying drug delivery system (SEDDS)[J]. Eur J Pharm Biopharm,2009,72(3):539-545. doi: 10.1016/j.ejpb.2009.03.001 [7] CHO H Y, CHOI J H, OH I J, et al. Self-emulsifying drug delivery system for enhancing bioavailability and lymphatic delivery of tacrolimus[J]. J Nanosci Nanotechnol,2015,15(2):1831-1841. doi: 10.1166/jnn.2015.9248 [8] 余越, 陶春, 杨海跃, 等. 不同孔径介孔二氧化硅纳米粒的制备及其用于固化西罗莫司自微乳[J]. 药学学报, 2017, 52(6):985-991. [9] HU X W, LIN C, CHEN D X, et al. Sirolimus solid self-microemulsifying pellets: formulation development, characterization and bioavailability evaluation[J]. Int J Pharm,2012,438(1-2):123-133. doi: 10.1016/j.ijpharm.2012.07.055 [10] 吴昊, 宋洪涛. 西罗莫司纳米结构脂质载体分散液的制备及其体外释放度考察[J]. 药学实践杂志, 2012, 30(3):189-193. doi: 10.3969/j.issn.1006-0111.2012.03.009 [11] 吴昊, 张晶, 宋洪涛. 西罗莫司纳米脂质载体固化制剂的制备[J]. 中国医药工业杂志, 2012, 43(7):562-567. doi: 10.3969/j.issn.1001-8255.2012.07.013 [12] 刘志宏, 胡雄伟, 张晶, 等. 西罗莫司固体自微乳化给药系统的体内外评价[J]. 中国新药杂志, 2016, 25(20):2369-2375. [13] 霍涛涛, 张美敬, 陶春, 等. 基于多组分评价的雷公藤提取物固体分散体的制备及体外表征[J]. 中草药, 2018, 49(1):128-134. doi: 10.7501/j.issn.0253-2670.2018.01.018 [14] SUN M H, SI L Q, ZHAI X Z, et al. The influence of co-solvents on the stability and bioavailability of rapamycin formulated in self-microemulsifying drug delivery systems[J]. Drug Dev Ind Pharm,2011,37(8):986-994. doi: 10.3109/03639045.2011.553618 [15] 侯明明, 宋洪涛, 周欣. 免疫抑制剂药物基因组学的研究进展[J]. 中国药房, 2009, 20(31):2471-2473. [16] 董文雪, 何军, 杨亚妮. 自微乳释药系统研究进展[J]. 中国医药工业杂志, 2011, 42(12):948-954. doi: 10.3969/j.issn.1001-8255.2011.12.020 [17] 刘建清, 张晶, 赵佳丽, 等. 纳米结构脂质载体促进难溶性药物口服吸收机制的研究进展[J]. 药学实践杂志, 2014, 32(4):254-256, 277. doi: 10.3969/j.issn.1006-0111.2014.04.004 [18] 王学功. 几种β-环糊精衍生物的合成及对药物分子的包结作用[D]. 天津: 天津大学, 2005. [19] 刘志宏, 胡雄伟, 陶春, 等. 西罗莫司自乳化给药系统及其固体化研究[J]. 药学实践杂志, 2016, 34(2):142-147, 170. doi: 10.3969/j.issn.1006-0111.2016.02.012 [20] GUMASTE S G, PAWLAK S A, DALRYMPLE D M, et al. Development of solid SEDDS, IV: effect of adsorbed lipid and surfactant on tableting properties and surface structures of different silicates[J]. Pharm Res,2013. doi: 10.1007/s11095-013-1114-4 [21] LEE D H, YEOM D W, SONG Y S, et al. Improved oral absorption of dutasteride via Soluplus®-based supersaturable self-emulsifying drug delivery system (S-SEDDS)[J]. Int J Pharm,2015,478(1):341-347. doi: 10.1016/j.ijpharm.2014.11.060 [22] 刘静. 自微乳释药系统在西药制剂中的应用[J]. 中国药业, 2018, 27(1):4-8. doi: 10.3969/j.issn.1006-4931.2018.01.002 [23] 徐晓琰, 恽菲, 狄留庆, 等. 环糊精包合药物胃肠道转运过程及机制研究进展[J]. 中草药, 2012, 43(10):2062-2065. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 6752
- HTML全文浏览量: 1995
- PDF下载量: 43
- 被引次数: 0
