

 下载:
下载:
 下载:
下载:

-
近年来,随着免疫抑制剂的广泛使用和介入手术的增多,深部真菌感染的发病率逐年上升。同时,手术患者的增加,住院天数的增长,也使得院内真菌感染成为急需解决的问题。在治疗真菌感染过程中长周期的给药方式和真菌耐药问题造成了政府和住院患者的医疗支出大幅增加,新的抗真菌药物或者协同抗真菌药物研究迫在眉睫。为了克服真菌的耐药性问题,本课题组一直致力于研究药物联用协同抗耐药真菌的效果。据统计,真菌感染中最常见菌属是念珠菌,而在念珠菌中白念珠菌的占比又高达45%~50%,因此,靶向于白念珠菌的药物研究就显得十分重要[1-6]。2010年,Morales等报道了化合物吩嗪硫酸甲酯具有抗白念珠菌被膜的活性[7]。为了进一步获得具有更强抗真菌活性的先导化合物,我们筛选了以吩嗪为骨架的20余种吩嗪类衍生物,获得了具有协同氟康唑抗真菌活性的化合物吩嗪衍生物-17,本研究为抗耐药真菌联合用药提供了新思路。
-
洁净工作台(HPeafe-1200LC(A2)上海力申科学仪器有限公司);振荡培养箱(HZ-2111K-B江苏太仓市实验设备厂);微量加样器(Biohit);小型冷冻离心机(HitachiCT15RE);蒸汽灭菌锅(KG-SX500 KAGOSHIMA SELSAKUSYO,Japan);多功能酶标仪(TECAN Infinite M200);倒置相差显微镜(Amersham Pharmacia AMG EVOS×1);紫外分光光度计(Amersham Biosciences Μltrospec10)。
-
白念珠菌标准菌株SC5314由美国Georgetown大学William A Fonzi教授赠送,临床分离的耐药白念珠菌103和538来自海军军医大学附属长海医院皮肤科,氟康唑(FCZ)由辉瑞公司生产,吩嗪类化合物购自chemdiv化合物库,二甲基亚砜(DMSO)购自博光生物试剂有限公司,酵母提取物、甘露醇、营养肉汤购自BD公司,蛋白胨、葡萄糖、琼脂均购自上海生工生物技术有限公司,RPMI1640购自Gibco公司。
-
将保存在SDA固体培养皿的SC5314单克隆菌株转接到1 ml YEPD培养基,30 ℃、200 r/min,培养过夜,使真菌菌株处于指数生长平台期。①洗菌:将活化好的菌株转移到1.5 ml离心管中,用无菌PBS缓冲液洗3次,再用1 ml PBS重悬。②调节浓度:用RPMI1640稀释菌液100倍,使其浓度为(1~5)×106 CFU/ml。再用RPMI1640稀释1000倍,使菌终浓度(1~5)×103 CFU/ml。③制备药敏实验板:取一块96孔板,第1列加入100 μl RPMI 1640液体培养基做空白对照;第2列加200 μl上述菌液,第3~12列分别加入100 μl上述菌液,第2列加吩嗪类衍生物使其终浓度为64 μg/ml,2~11列进行倍比稀释,使得药物终浓度分别为64、32、16、8、4、2、1、0.5、0.25、0.125 μg/ml,每个药都加2个复孔(2行),以减少实验误差,铺好药和菌的96孔板用封条封闭,于30 ℃恒温培养过夜,用酶标仪在λ=630 nm测吸光度(A)值,计算MIC80值。
-
菌株活化、洗菌和调菌浓度同前所述,取一块96孔板,第1列加入100 μl RPMI1640/YEPD培养基做空白对照;第12列加入100 μl上述菌液做阳性对照;剩余菌液加入适量氟康唑,使得氟康唑终浓度为2 μg/ml,第2列加入该配制好的菌液200 μl,第3~11列加入该配制好的菌液100 μl,第2列再加入吩嗪类衍生物使其终浓度为64 μg/ml,2~11列进行倍比稀释,使得药物终浓度分别为64、32、16、8、4、2、1、0.5、0.25、0.125 μg/ml,每种药都加2个复孔(2行),以减少实验误差,将铺好药和菌的96孔板用封条封闭,于30 ℃恒温培养过夜,用酶标仪在λ=630 nm处测A值,计算最低抑菌浓度(MIC80)和协同指数(FICI)。
除了用上述耐药白念珠菌103实验外,再更换菌株为538,重复上述实验步骤进行实验。
-
过夜活化的白念珠菌SC5314分别用Spider培养液和RPMI1640培养液稀释1 000倍,分别加药使吩嗪衍生物-17终浓度为4 μg/ml、FCZ的终浓度为4 μg/ml以及吩嗪衍生物-17与FCZ的合用(浓度与单药相同),空白对照加入同体积的DMSO,充分混匀,转移至12孔板中,37 ℃,静置培养3 h,倒置显微镜观察菌丝形态。
-
通过对吩嗪类24种衍生物的体外抗真菌活性研究发现,吩嗪类化合物单用对白念珠菌没有抗真菌活性,其MIC80均>128 μg/ml。在RPMI1640培养液中,采用临床分离的氟康唑耐药白念珠菌103和538(氟康唑单用时,MIC80>32 μg/ml),考察吩嗪类化合物协同氟康唑的抗真菌活性。结果显示,当吩嗪类化合物与2 μg/ml的氟康唑合用时,0.25~0.5 μg/ml的吩嗪衍生物-12、0.125 μg/ml的吩嗪衍生物-17、1~2 μg/ml的吩嗪衍生物-18协同氟康唑后有明显的抗耐药白念珠菌的活性,结果见表1。而后我们采用棋盘式微量液基稀释法,进一步考察了氟康唑和吩嗪衍生物-12、吩嗪衍生物-17、吩嗪衍生物-18合用时的最低浓度,结果显示吩嗪衍生物-17与氟康唑协同抗白念珠菌菌效果最强,0.0625 μg/ml的吩嗪衍生物-17与1 μg/ml的氟康唑即可完全抑制耐药白念珠菌的生长,协同指数FICI<0.5,说明与氟康唑具有明显的协同作用(表2)。
表 1 吩嗪类衍生物(2 μg/ml)协同氟康唑对氟康唑耐药白念珠菌103MIC80值的测定结果
化合物编号 结构 分离株103MIC80
(μg/ml)分离株538MIC80
(μg/ml)吩嗪-1 >16 >16 吩嗪-2 8 8 吩嗪-3 >16 16 吩嗪-4 >16 >16 吩嗪-5 >16 >16 吩嗪-6 8 8 吩嗪-7 >16 >16 吩嗪-8 16 8 吩嗪-9 >16 >16 吩嗪-10 >16 >16 吩嗪-11 16 16 吩嗪-12 0.5 0.25 吩嗪-13 >16 >16 吩嗪-14 >16 >16 吩嗪-15 >16 >16 吩嗪-16 >16 >16 吩嗪-17 0.125 0.125 吩嗪-18 2 1 吩嗪-19 8 8 吩嗪-20 16 8 吩嗪-21 16 16 吩嗪-22 >16 >16 吩嗪-23 >16 >16 吩嗪-24 >16 16 表 2 吩嗪类衍生物与氟康唑单用及合用对氟康唑耐药白念珠菌538的MIC80值和FICI值的测定结果
化合物编号 单用[MIC80(μg/ml)] 合用[MIC80(μg/ml)] FICI 协同方式 吩嗪类衍生物 氟康唑 吩嗪类衍生物 氟康唑 吩嗪类衍生物-12 >64 >32 0.25 1.0 0.034 协同 吩嗪类衍生物-17 >64 >32 0.0625 1.0 0.032 协同 吩嗪类衍生物-18 >64 >32 0.5 2.0 0.070 协同 -
为了进一步考察吩嗪类衍生物-17的体外抗菌活性,我们考察了其对菌丝生长的抑制作用。结果表明,在RPMI1640诱导菌丝形成的过程中,吩嗪衍生物-17与氟康唑合用对菌丝的生长抑制不明显,但在Spider培养基中,两药合用对菌丝的形成有明显抑制作用(图1)。
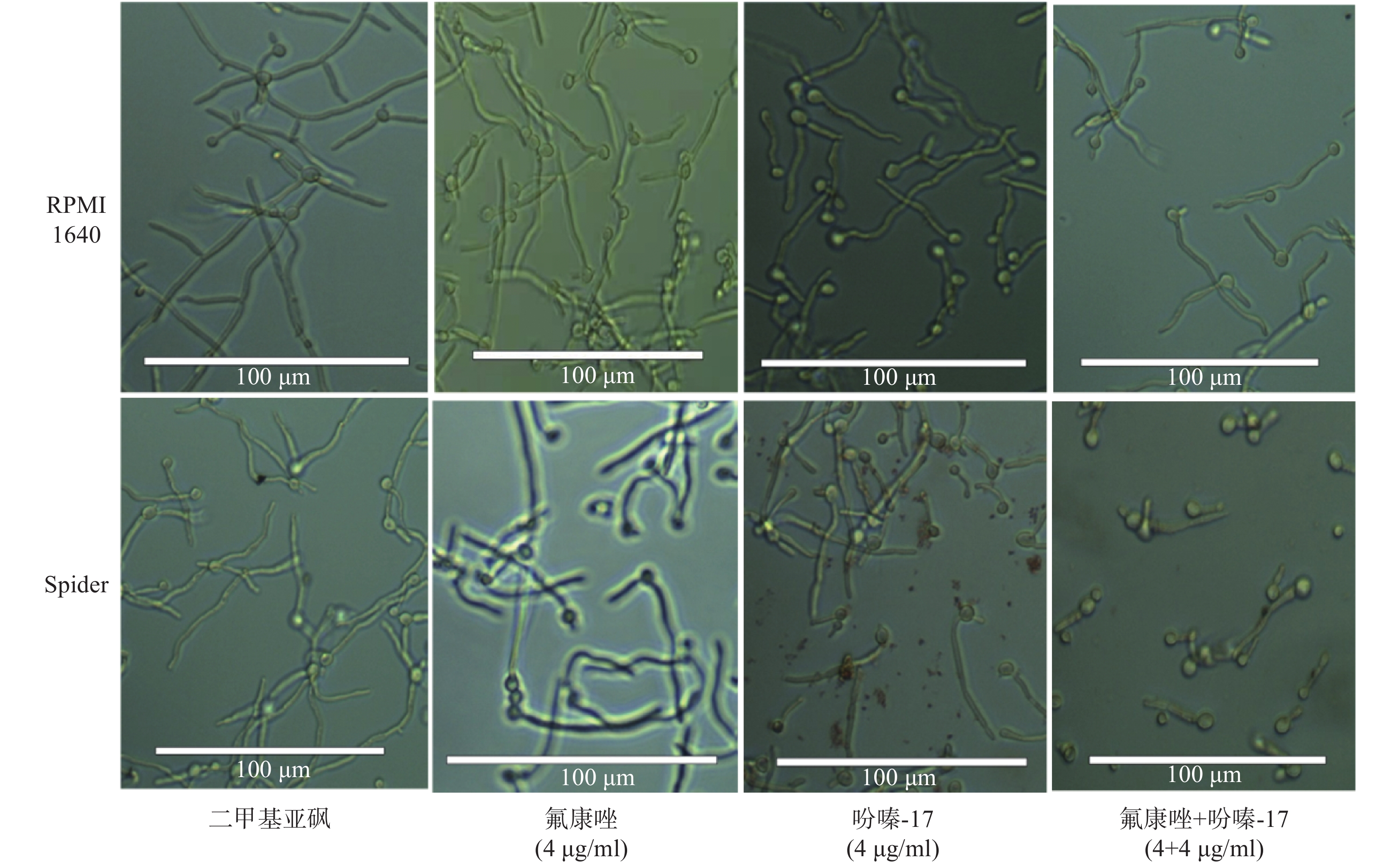
图 1 吩嗪类衍生物-17抑制白念珠菌的菌丝形成
-
近年来,随着免疫抑制剂的广泛用、手术介入和免疫缺陷患者的增多,系统性真菌感染的发病率逐年攀升。但抗真菌药物的研发进展缓慢,现有的几类抗真菌药物在长期使用后也逐渐出现了耐药性。为克服临床真菌感染的问题,研发新的抗真菌药物意义重大。本课题组长期致力于协同抗耐药真菌研究,期望筛选获得具有协同抗耐药真菌活性的化合物,进而达到降低给药剂量,提高抗菌能力的目的,为临床抗真菌治疗提供新的策略。本研究前期通过体外筛选吩嗪类化合物的抗真菌活性,发现吩嗪类化合物单药使用时没有抗真菌活性,但是吩嗪衍生物-12、-17、-18与氟康唑合用有显著的协同抗真菌活性,这一研究结果有望为抗真菌药物研发提供新的思路。该研究尚未探讨吩嗪类衍生物的协同抗耐药真菌机制,后续仍需进一步深入研究。
Study on the antifungal activity of phenazine derivatives
-
摘要:
目的 研究吩嗪类衍生物的抗真菌活性。 方法 利用微量液基稀释法考察吩嗪类衍生物的体外抗真菌活性;利用棋盘式微量稀释法检测吩嗪类衍生物与氟康唑合用对常见临床耐药菌的抗真菌活性;在菌丝诱导条件下考察吩嗪衍生物对白念珠菌菌丝形成的抑制效果。 结果 吩嗪类衍生物单用对临床常见条件致病真菌白念珠菌没有明显抗真菌活性;吩嗪衍生物-17与氟康唑合用有显著抗白念珠菌活性;吩嗪衍生物-17与氟康唑合用可以明显抑制白念珠菌菌丝生长。 结论 筛选获得了与氟康唑合用具有抗真菌活性的吩嗪类衍生物-17,为抗真菌药物研发和克服真菌耐药提供了新思路。 Abstract:Objective To study the antifungal activity of phenazines derivatives. Methods The anti-fungal activity of phenazine compounds was evaluated initially with micro-liquid dilution. No significant antifungal activity against Candida albicans was found. Then, with the combination of phenazine compounds and fluconazole, the anti-fungal activity against fluconazole-resistant C. albicans was detected. Results The phenazine-17 had significant antifungal activity when combined with fluconazole through the inhibition of hyphae formation. Conclusion This study provides a new idea for the development of antifungal drugs and the solution of antifungal drug resistance. -
Key words:
- phenazine derivatives /
- Candida albicans /
- antifungal activity /
- hyphae formation
-
放线菌以其能产生结构新颖且有良好生物活性的先导化合物而备受关注[1],一直被认为是天然药物的重要生产者,其主要结构类型包括聚酮、生物碱、多肽和萜烯类化合物等,同时涵盖了多种多样的生物活性如抗菌、抗寄生虫、免疫调节、抗炎、抗癌等[2-4],这突显了放线菌具有不可预估的药物开发潜力。
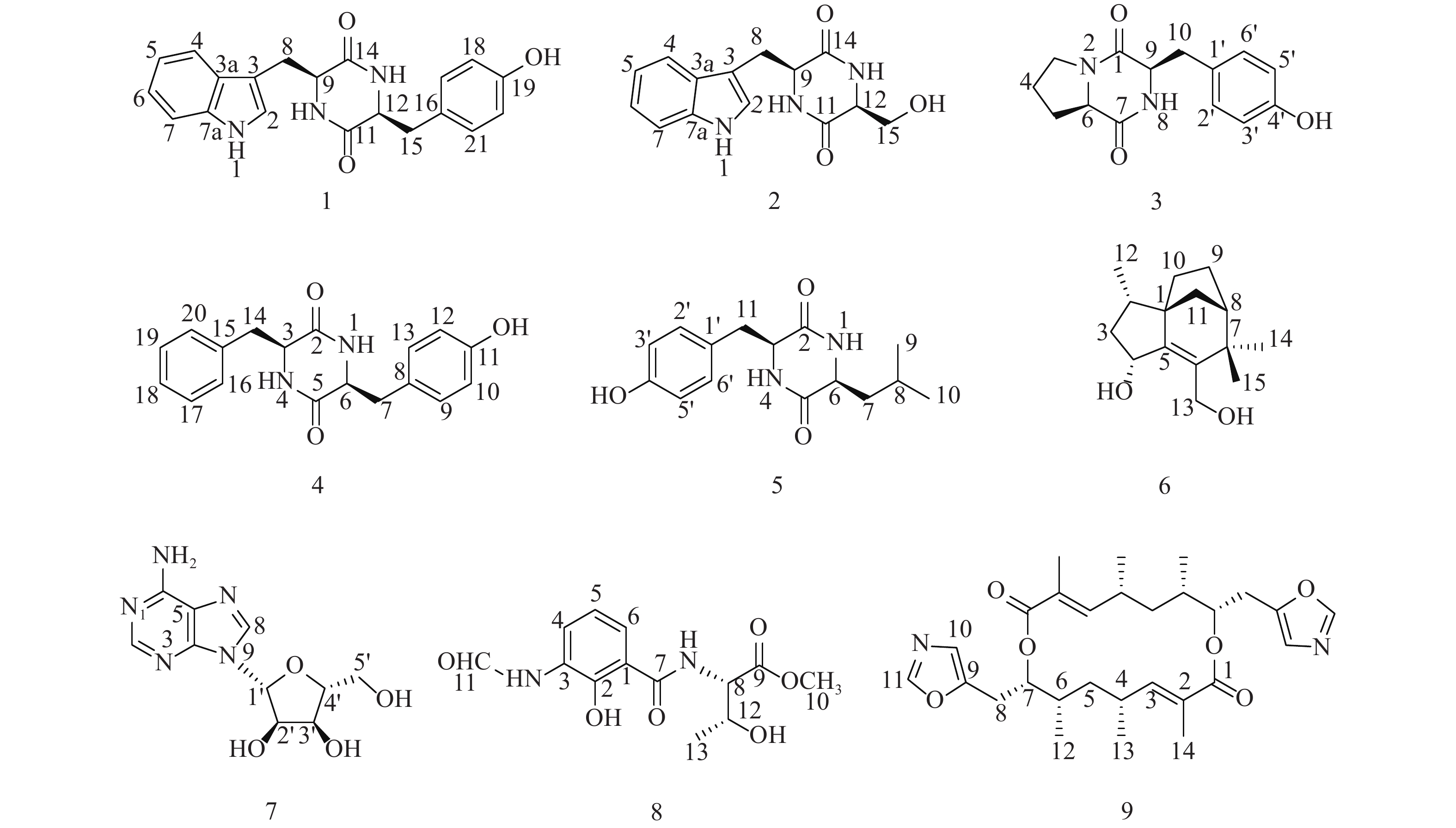
随着研究的深入,陆地和普通环境中的资源日趋枯竭,很多微生物及其次生代谢产物被重复开发和提取分离,发现新活性分子的几率愈来愈低,开发创新药物的难度越来越大[5-6];而极地极端的生态环境造就的微生物具有产生更为特别的化学骨架和活性次生代谢产物的能力,是新型药源分子的重要来源。本文以采自北极楚克奇海域海绵共附生放线菌Streptomyces sp. LHW11-07为研究对象,从其发酵浸膏中分离鉴定了9个单体化合物1~9(图1),其中化合物1和2为该属内首次分离得到。
1. 材料和方法
1.1 实验仪器与试剂
AMX-600型核磁共振仪(德国Bruker公司);Xevo G2-XS Q-TOF液质联用仪、1525/2996, 2998型高效液相色谱仪(美国Waters公司);半制备型HPLC色谱柱(Atlantis Prep T3,美国Waters公司;YMC C18,日本YMC公司);中压柱色谱仪(法国Interchim公司);恒温振荡培养箱(上海一恒科学仪器有限公司);N-1000型旋转蒸发仪(上海爱郎仪器有限公司);反相ODS硅胶和Sephadex LH-20柱色谱填料(Pharmacia公司);正相硅胶(200-300目)和TLC薄层板(烟台江友硅胶开发有限公司);分析级试剂(上海化学试剂公司);色谱级试剂(德国Merck公司);氘代试剂(美国剑桥同位素实验室公司)。
1.2 菌株的来源及鉴定
菌株分离于北极海域来源的海绵样本,经16S rRNA基因序列鉴定为Streptomyces sp.,编号为LHW11-07,菌种保存于上海交通大学医学院附属仁济医院药学部海洋药物研究中心。
1.3 菌株的大发酵
培养基为ISP2:葡萄糖(4 g/L)、酵母提取物(4 g/L)、麦芽糖提取物(10 g/L)以及海盐(25 g/L),加水溶解后调节pH为7.2~7.4,分装后高压灭菌20 min (121 ℃),冷却备用。
挑取Streptomyces sp. LHW11-07单菌落至1级种子培养基里(100 ml ISP2培养基至250 ml三角瓶),置于30 ℃,220 r/min的恒温摇床培养3 d,得1级种子液;将1级种子液按5%接种量接到2级种子培养基里(150 ml ISP2培养基至500 ml三角瓶),置于30 ℃,220 r/min的恒温摇床培养3 d,得2级种子液;将2级种子液按5%接种量接到大发酵培养基里(700 ml ISP2培养基至2 L三角瓶),置于30 ℃,220 r/min的恒温摇床培养7 d,共得到发酵液50 L。
1.4 发酵产物的提取与分离
菌株培养7 d后,用等体积的乙酸乙酯萃取3次,合并乙酸乙酯提取液,减压浓缩得粗浸膏9.8 g。粗浸膏先经凝胶柱分离,二氯甲烷:甲醇(1∶1)的混合溶剂进行洗脱,得到组份Fr.1~Fr.7。
组份Fr.4经正相中压柱色谱分离(二氯甲烷:甲醇100:0~0:100),得到组份Fr.4a~Fr.4j。组份Fr.4d再经反相中压柱色谱分离(10%~100%乙腈水),得到组份Fr.4d1~Fr.4d8,Fr.4d2和Fr.4d3用反相半制备HPLC纯化(35%甲醇水,YMC C18),分别得到化合物1 (12 mg, tR=26 min)和2 (47.6 mg, tR=30 min);Fr.4d6用反相半制备HPLC纯化(49%甲醇水,YMC C18),得到化合物3 (8.4 mg, tR=23 min)和4 (2.0 mg, tR=30 min);组份Fr.4g再经凝胶柱纯化,洗脱剂为正己烷:二氯甲烷:甲醇(4∶5∶1),得到组份Fr.4g1~Fr.4g11,其中Fr.4g3用反相半制备HPLC纯化(25%乙腈水,YMC C18),得到化合物5 (1.2 mg, tR=18 min)。
组份Fr.7经反相中压柱色谱分离(10%~100%乙腈水),得到组份Fr.7A~Fr.7D。组份Fr.7A用反相半制备HPLC纯化(20%乙腈水,YMC C18),得到化合物6 (18 mg, tR=19 min)和7 (3.4 mg, tR=25 min);组份Fr.7B经正相中压柱分离(石油醚:丙酮100:0~0:100),得到组份Fr.7B1~Fr.7B9,Fr.7B4用反相半制备HPLC纯化(88%乙腈水,Atlantis Prep T3),得到化合物8 (12 mg, tR=23 min)和9 (8.4 mg, tR=26 min)。
2. 结构鉴定
化合物1:淡黄色固体,ESI-MS显示准分子离子峰m/z 372 [M+Na]+。1H-NMR (600 MHz, DMSO)显示在低场区有吲哚环的特征信号δH 7.00 (1H, s, H-2),7.49 (1H, d, J=8.0 Hz, H-4),7.02 (1H, t, J=7.6 Hz, H-5),7.05 (1H, t, J=7.6 Hz, H-6),7.32 (1H, d, J=8.0 Hz, H-7);4个活泼氢质子信号δH 10.89 (1H, d, J=2.6 Hz, NH-1),7.83 (1H, d, J=3.0 Hz, NH-10),7.62 (1H, d, J=3.0 Hz, NH-13),9.20 (1H, s, OH-19);4个芳香质子信号δH 6.53 (2H, d, J=8.4 Hz, H-17, H-21),6.59 (2H, d, J=8.4 Hz, H-18, H-20),提示分子中有1个对位二取代的苯环;在高场区有两组亚甲基质子信号δH 2.80 (1H, dd, J=14.5, 4.5 Hz, H-8),2.43 (1H, ov, H-8),δH 1.83 (1H, dd, J=13.4, 6.9 Hz, H-15),2.47 (1H, ov, H-15),两个次甲基质子信号δH 4.01 (1H, m, H-9),3.95 (1H, m, H-12)。13C-NMR (150 MHz, DMSO)结合DEPT谱表明其有20个碳信号,2个酮羰基碳δC 166.7,166.2,14个芳香碳,2个亚甲基碳δC 30.0,40.0和2个次甲基碳δC 55.9,55.2。对其碳信号进行归属:δC 118.7 (C-2)、108.9 (C-3)、127.5 (C-3a)、118.4 (C-4)、120.8 (C-5)、124.3 (C-6)、111.3 (C-7)、136.0 (C-7a)、30.0 (C-8)、55.9 (C-9)、166.7 (C-11)、55.2 (C-12)、166.2 (C-14)、40.1 (C-15)、126.4 (C-16)、130.7 (C-17, C-21)、114.9 (C-18, C-20)、156.0 (C-19)。以上数据与文献[7]对比基本一致,故确定为cyclo-(L-Tyr-L-Trp)。
化合物2:淡黄色固体,ESI-MS显示准分子离子峰m/z 274 [M+H]+。1H-NMR (600 MHz, DMSO)发现其与化合物1一样有吲哚环的特征信号δH 7.09 (1H, s, H-2),7.52 (1H, d, J = 7.9 Hz, H-4),6.99 (1H, t, J = 7.8 Hz, H-5),7.02 (1H, t, J = 7.8 Hz, H-6),7.30 (1H, d, J = 7.8 Hz, H-7);3个活泼氨基质子信号δH 10.87 (1H, s, NH-1),δH 8.30 (1H, m, NH-10),7.85 (1H, d, J = 2.9 Hz, NH-13);两组亚甲基质子信号δH 3.21 (1H, m, H-8),3.13 (1H, m, H-8),δH 3.65 (1H, m, H-15),3.05 (1H, m, H-15),两个次甲基质子信号δH 4.87 (1H, m, H-9),4.00 (1H, m, H-12)。13C-NMR (150 MHz, DMSO)结合DEPT谱表明其有14个碳信号,2个酮羰基碳δC 167.2,165.7,8个芳香碳,2个亚甲基碳δC 63.0,30.3和2个次甲基碳δC 57.3,55.5。对其碳信号进行归属:δC 127.6 (C-2)、111.2 (C-3)、136.0 (C-3a)、118.6 (C-4)、120.8 (C-5)、124.0 (C-6)、118.3 (C-7)、109.0 (C-7a)、30.3 (C-8)、57.3 (C-9)、167.2 (C-11)、55.5 (C-12)、165.7 (C-14)、63.0 (C-15)。以上数据与文献[8]对比基本一致,故确定为cyclo-(L-Trp-L-Ser)。
化合物3:白色固体,ESI-MS显示准分子离子峰m/z 261 [M+H]+。1H-NMR (600 MHz, DMSO)提示有2个活泼氢质子信号δH 7.87 (1H, s, NH-8),9.22 (1H, s, OH-4’);1组对位二取代的苯环芳香质子信号δH 7.04 (2H, d, J = 8.2 Hz, H-2’, H-6’),6.63 (2H, d, J = 8.2 Hz, H-3’, H-5’);2个次甲基质子信号δH 4.24 (1H, t, J = 8.2 Hz, H-6),4.03 (1H, dd, J = 9.9, 2.9 Hz, H-9);4组亚甲基质子信号δH 3.42 (1H, m, H-3),3.24 (1H, m, H-3),1.73 (2H, m, H2-4),2.00 (1H, m, H-5),1.41 (1H, m, H-5),2.93 (2H, m, H2-10)。13C-NMR (150 MHz, DMSO)结合DEPT谱表明其有14个碳信号,2个酮羰基碳δC 168.9,165.1,6个芳香碳,2个次甲基碳δC 58.4,56.0以及4个亚甲基碳δC 44.6,34.7,27.8,21.9。对其碳信号进行归属:δC 165.1 (C-1)、44.6 (C-3)、21.9 (C-4)、27.8 (C-5)、58.4 (C-6)、168.9 (C-7)、56.0 (C-9)、34.7 (C-10)、127.0 (C-1’)、130.8 (C-2’, C-6’)、114.8 (C-3’, C-5’)、155.9 (C-4’)。以上数据与文献[9]对比基本一致,故确定为cyclo-(D-Tyr-D-Pro)。
化合物4:白色固体,ESI-MS显示准分子离子峰m/z 311 [M+H]+。1H-NMR (600 MHz, DMSO)显示3个活泼氢质子信号δH 9.30 (1H, s, OH-11),7.84 (2H, t, J = 2.9 Hz, NH-1, NH-4);9个芳香区质子信号:4个归为1组对位二取代苯环δH 6.85 (2H, d, J = 8.5 Hz, H-9, H-13),6.65 (2H, t, J = 8.5 Hz, H-10, H-12),5个归为1组单取代苯环δH 7.20 (1H, t, J = 7.6 Hz, H-18),7.04 (2H, d, J = 6.9 Hz, H-16, H-20),7.28 (2H, t, J = 7.6 Hz, H-17, H-19);2组亚甲基质子信号δH 2.58 (1H, dd, J = 13.6, 5.0 Hz, H-7),2.20 (1H, d, J = 6.5 Hz, H-7),2.19 (2H, dd, J = 13.6, 6.5 Hz, H2-14),2个次甲基质子信号δH 3.95 (1H, m, H-3),3.90 (1H, m, H-6)。13C-NMR (150 MHz, DMSO)结合DEPT谱显示其有18个碳信号,2个酮羰基碳δC 166.2,166.2,12个芳香碳,2个亚甲基碳δC 40.1,38.5和2个次甲基碳δC 55.7,55.4。对其碳信号进行归属:δC 166.2 (C-2)、55.7 (C-3)、166.3 (C-5)、55.4 (C-6)、40.1 (C-7)、126.5 (C-8)、130.8 (C-9, C-13)、115.0 (C-10, C-12)、156.1 (C-11)、38.5 (C-14)、136.7 (C-15)、129.7 (C-16, C-20)、128.2 (C-17, C-19)、126.4 (C-18)。以上数据与文献[10]对比基本一致,故确定为cyclo-(L-Tyr-L-Phe)。
化合物5:白色固体,ESI-MS显示准分子离子峰m/z 277 [M+H]+。1H-NMR (600 MHz, DMSO)显示3个活泼氢质子信号δH 9.22 (1H, s, OH-4’),8.02 (2H, dd, J = 5.6, 2.5 Hz, NH-1, NH-4);4个芳香质子信号δH 6.90 (2H, d, J = 8.2 Hz, H-2’, H-6’),6.64 (2H, d, J = 8.2 Hz, H-3’, H-5’),提示分子中有1个对位二取代苯环;3个次甲基质子信号δH 4.06 (1H, q, J = 3.3 Hz, H-3),3.44 (1H, m, H-6),1.43 (1H, ov, H-8),2个亚甲基质子信号δH 1.43 (1H, m, H-7),1.23 (1H, m, H-7),2.69 (1H, q, J = 13.6, 4.8 Hz, H-11),3.01 (1H, q, J = 13.7, 3.7 Hz, H-11)以及2个末端甲基质子信号δH 0.63 (6H, ov, H3-9, H3-10)。13C-NMR (150 MHz, DMSO)结合DEPT谱显示其有15个碳信号,2个酮羰基碳δC 166.2,167.4,6个芳香碳,2个亚甲基碳δC 43.7,37.7,3个次甲基碳δC 55.7,52.3,21.4以及2个甲基碳δC 22.9,22.8。对其碳信号进行归属:δC 166.2 (C-2)、55.7 (C-3)、167.4 (C-5)、52.3 (C-6)、43.7 (C-7)、21.4 (C-8)、22.9 (C-9)、22.8 (C-10)、37.7 (C-11)、125.8 (C-1’)、131.2 (C-2’, C-6’)、114.8 (C-3’, C-5’)、156.4 (C-4’)。以上数据与文献[11]对比基本一致,故确定为cyclo-(L-Tyr-L-Leu)。
化合物6:白色固体,ESI-MS显示准分子离子峰m/z 259 [M+Na]+。1H-NMR (600 MHz, DMSO)显示有2个活泼氢质子信号δH 5.05 (1H, d, J = 4.5 Hz, OH-4)和4.39 (1H, q, J = 4.0 Hz, OH-13),3个甲基质子信号δH 0.88 (3H, d, J = 6.8 Hz, H3-12),0.98 (3H, s, H3-14)和δH 1.05 (3H, s, H3-15),5对亚甲基质子信号δH 2.14 (1H, m, H-3),1.23 (1H, m, H-3),1.74 (1H, m, H-9),1.60 (1H, m, H-9),1.44 (3H, m, H2-10, H-11),1.32 (1H, d, J = 10.4 Hz, H-11),3.95 (2H, m, H2-13),3个次甲基质子信号δH 1.68 (1H, m, H-2),4.57 (1H, m, H-4),1.77(1H, m, H-8)。13C-NMR (150 MHz, DMSO)结合DEPT谱共显示有15个碳信号,包括4个季碳δC 52.1,150.1,136.7,39.8;3个次甲基碳δC 35.2,69.8,46.4;5个亚甲基碳δC 42.4,23.8,28.7,36.4,57.1以及3个甲基碳δC 13.7,29.1,24.4。对其碳信号进行归属:δC 52.1 (C-1)、35.2 (C-2)、42.4 (C-3)、68.9 (C-4)、150.1 (C-5)、136.7 (C-6)、39.8 (C-7)、46.4 (C-8)、23.8 (C-9)、28.7 (C-10)、36.4 (C-11)、13.7 (C-12)、57.1 (C-13)、29.1 (C-14)、24.4 (C-15)。以上数据与文献[12]对比基本一致,故确定为albaflavenol B。
化合物7:白色结晶固体,ESI-MS显示准分子离子峰m/z 268 [M+H]+。1H-NMR (600 MHz, DMSO)可看出其有13个氢信号,包括5个活泼氢质子信号δH 3.56 (2H, m, NH2),5.49 (1H, s, OH-5’),5.36 (1H, t, J = 4.9 Hz, OH-2’)和5.23 (1H, s, OH-3’),4个连氧次甲基质子信号δH 4.56 (1H, s, H-2’),4.14 (1H, s, H-3’),3.96 (1H, m, H-4’)和5.90 (1H, d, J = 5.8 Hz, H-1’),1组亚甲基信号δH 3.66 (2H, m, H2-5’)以及2个低场区的烯氢质子信号δH 8.37 (1H, s, H-8)和8.21 (1H, s, H-2)。13C-NMR (150 MHz, DMSO)显示其共有10个碳信号,结合DEPT谱可推测有3个芳香季碳δC 149.9,119.8,154.3,2个连氮的芳香次甲基碳δC 151.7,138.6,4个次甲基碳δC 87.8,73.5,70.5,85.7,1个亚甲基碳δC 61.5。对其碳信号进行归属:δC 151.7 (C-2)、149.9 (C-4)、119.8 (C-5)、154.3 (C-6)、138.6 (C-8)、87.8 (C-1’)、73.5 (C-2’)、70.5 (C-3’)、85.7 (C-4’)、61.5 (C-5’)。以上数据与文献[13]对比基本吻合,故确定为β-adenosine。
化合物8:绿色无定型固体,ESI-MS显示准分子离子峰m/z 297 [M+H]+。1H-NMR (600 MHz, MeOD)显示有12个氢信号,包括1个活泼氢质子信号δH 8.37 (1H, s, H-11),1个甲氧基质子信号δH 3.79 (3H, s),1个甲基质子信号δH 1.25 (3H, d, J = 6.4 Hz, H3-13),3个低场区的芳香氢质子信号δH 8.31 (1H, d, J = 7.8 Hz, H-4),6.92 (1H, t, J = 8.0 Hz, H-5)和7.65 (1H, d, J = 8.0 Hz, H-6),以及2个次甲基质子信号δH 4.74 (1H, d, J = 3.2 Hz, H-8)和4.40 (1H, m, H-12),后与文献[14]对比发现其有4个活泼氢质子信号没有显示出来,而根据相关化学位移可确定其是同一个已知化合物。13C-NMR (150 MHz, MeOD)显示有13个碳信号,结合DEPT谱可推测有6个芳香碳δC 123.4,119.4,126.2,128.2,152.5,115.6,2个羰基碳δC 171.8,172.4,而δC 162.1为醛基碳,2个次甲基碳δC 59.4,68.4,1个甲基碳δC 20.5,以及1个甲氧基碳δC 52.9。对其碳信号进行归属:δC 115.6 (C-1)、152.5 (C-2)、128.2 (C-3)、126.2 (C-4)、119.4 (C-5)、123.4 (C-6)、171.8 (C-7)、59.4 (C-8)、172.4 (C-9)、52.9 (C-10)、162.1 (C-11)、68.4 (C-12)、20.5 (C-13)。以上数据与文献[14]对比基本吻合,故确定为N-formylantimyic acid methyl ester。
化合物9:白色粉末状固体,ESI-MS显示准分子离子峰m/z 499 [M+H]+。1H-NMR (600 MHz, DMSO)显示有19个氢信号:δH 8.20 (1H, s, H-11),6.82 (1H, s, H-10),6.32 (1H, dd, J = 10.5, 1.3 Hz, H-3),5.01 (1H, m, H-7),2.99 (1H, dd, J = 2.5, 15.8 Hz, H-8),2.80 (1H, dd, J = 10.3, 15.6 Hz, H-8),2.58 (1H, m, H-4),1.66 (1H, m, H-5),1.65 (3H, s, 2-Me),1.27 (1H, m, H-6),1.25 (1H, m, H-5),1.06 (3H, d, J = 6.5 Hz, 4-Me)和0.95 (3H, d, J = 5.9 Hz, 6-Me)。而13C-NMR (150 MHz, DMSO)结合DEPT谱显示只有14个碳信号:δC 166.0 (C-1),126.8 (C-2),147.4 (C-3),30.7 (C-4),37.6 (C-5),35.1 (C-6),74.5 (C-7),24.0 (C-8),149.4 (C-9),122.9 (C-10),151.3 (C-11),12.7 (2-Me),21.1 (4-Me)和16.2 (6-Me),说明这个化合物可能是一个具有对称结构的二聚体,通过与文献[15]中化合物conglobatin A对比后发现两者波谱数据完全吻合,故最终确定为conglobatin A。
3. 讨论
自上世纪发现青霉素以来,微生物中活性次生代谢产物一直是药物先导化合物的重要来源之一,据统计1940年—2019年间,科学家从微生物中开发出293种治疗不同疾病的临床药物[16]。但随着研究的深入,很多微生物及其次生代谢产物存在被重复开发和提取分离的问题,加之多重耐药性的产生,迫使人们需要开拓新的制造药物的微生物来源[5-6],而其中极地微生物资源是珍贵而特殊的。来自极地海洋等特殊生态环境的生物往往具有比陆地生物更为丰富的代谢途径和功能基因簇,增加了产生结构新颖且功能独特的次生代谢物的可能性。极地生物以微生物和一些能适应极端条件的海洋生物为主,然而与已报道的大量极地微生物相比,鲜有微生物活性天然产物相关研究报道,因此,极地微生物极具研究价值[17-18]。
笔者以一株采自北极海域海绵共附生放线菌Streptomyces sp. LHW11-07为研究对象,从其发酵浸膏中分离得到9个单体化合物1~9,包括环二肽化合物1~5,倍半萜化合物6,核苷类化合物7,以及两个其他结构类型化合物8和9,其中化合物1和2是首次分离于Streptomyces放线菌,而这些化合物的生物活性还有待进一步探究;本研究进一步丰富了该属放线菌的化学多样性,同时,为高值化开发利用极地微生物这一国家战略资源提供了物质基础和理论依据。
据文献报道,化合物1对所测试的病原性细菌和真菌均具有一定的对抗作用[19],化合物2测试了4种肿瘤细胞均无明显的细胞毒性[20],化合物3对海胆Strongylocentrotus intermedius胚胎具有细胞毒活性[9],化合物5具有抗炎活性并对H1N1和RSV病毒有一定的杀伤作用[21],化合物7作为一种内源性嘌呤核苷,具有降低血压、抑制血小板聚焦、舒张血管、减慢心律等生理活性[22],而化合物9可抑制癌细胞株的增殖,在体外对Trypanosoma brucei brucei GUTat 3.1表现出抗锥虫体活性等[23]。
-
图 1 吩嗪类衍生物-17抑制白念珠菌的菌丝形成
注:白念珠菌在37 ℃的RPMI 1640或Spider培养基中诱导3 h,氟康唑和吩嗪衍生物-17加入的浓度均为4 μg/ml。
表 1 吩嗪类衍生物(2 μg/ml)协同氟康唑对氟康唑耐药白念珠菌103MIC80值的测定结果
化合物编号 结构 分离株103MIC80
(μg/ml)分离株538MIC80
(μg/ml)吩嗪-1 >16 >16 吩嗪-2 8 8 吩嗪-3 >16 16 吩嗪-4 >16 >16 吩嗪-5 >16 >16 吩嗪-6 8 8 吩嗪-7 >16 >16 吩嗪-8 16 8 吩嗪-9 >16 >16 吩嗪-10 >16 >16 吩嗪-11 16 16 吩嗪-12 0.5 0.25 吩嗪-13 >16 >16 吩嗪-14 >16 >16 吩嗪-15 >16 >16 吩嗪-16 >16 >16 吩嗪-17 0.125 0.125 吩嗪-18 2 1 吩嗪-19 8 8 吩嗪-20 16 8 吩嗪-21 16 16 吩嗪-22 >16 >16 吩嗪-23 >16 >16 吩嗪-24 >16 16  下载: 导出CSV
下载: 导出CSV
表 2 吩嗪类衍生物与氟康唑单用及合用对氟康唑耐药白念珠菌538的MIC80值和FICI值的测定结果
化合物编号 单用[MIC80(μg/ml)] 合用[MIC80(μg/ml)] FICI 协同方式 吩嗪类衍生物 氟康唑 吩嗪类衍生物 氟康唑 吩嗪类衍生物-12 >64 >32 0.25 1.0 0.034 协同 吩嗪类衍生物-17 >64 >32 0.0625 1.0 0.032 协同 吩嗪类衍生物-18 >64 >32 0.5 2.0 0.070 协同
下载: 导出CSV
-
[1] 廖万清. 深部真菌感染治疗进展[J]. 中国麻风皮肤病杂志, 2003, 19(6):597-600. doi: 10.3969/j.issn.1009-1157.2003.06.036 [2] FRIDKIN SK, JARVIS WR. Epidemiology of nosocomial fungal infections[J]. Clin Microbiol Rev,1996,9(4):499-511. [3] PFALLER MA. Nosocomial candidiasis: emerging species, reservoirs, and modes of transmission[J]. Clin Infect Dis,1996,22(Suppl 2):S89-S94. [4] LEE JS, JUNG WK, JEONG MH, et al. Sanguinarine induces apoptosis of HT-29 human colon cancer cells via the regulation of Bax/Bcl-2 ratio and caspase-9-dependent pathway[J]. Int J Toxicol,2012,31(1):70-77. doi: 10.1177/1091581811423845 [5] SANDAI D, TABANA YM, OUWEINI AE, et al. Resistance of Candida albicans biofilms to drugs and the host immune system[J]. Jundishapur J Microbiol,2016,9(11):e37385. [6] HARVEY AL, EDRADA ER, QUINN RJ. The re-emergence of natural products for drug discovery in the genomics era[J]. Nat Rev Drug Discov,2015,14(2):111-129. doi: 10.1038/nrd4510 [7] MORALES DK, JACOBS NJ, RAJAMANI S, et al. Antifungal mechanisms by which a novel Pseudomonas aeruginosa phenazine toxin kills Candida albicans in biofilms[J]. Mol Microbiol,2010,78(6):1379-1392. doi: 10.1111/j.1365-2958.2010.07414.x -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4467
- HTML全文浏览量: 1633
- PDF下载量: 32
- 被引次数: 0
