

 下载:
下载:
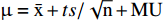
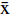
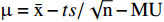
 下载:
下载:



-
小檗碱是从毛茛科植物根茎中提取的一种季胺类异喹啉类生物碱,又称黄连素,为中药黄连的主要成分,临床多用其盐酸盐。盐酸小檗碱(BBR)的药理作用主要为清热解毒,属于传统的消化系统用药,临床上常用作抗菌剂,用于治疗细菌性痢疾、肠胃炎等肠道感染[1-2]。盐酸小檗碱溶解度低, 渗透性差,在口服给药时,还存在药物释放快、半衰期短、生物利用度低等缺陷。对于需要达到一定血药浓度为疗效前提的疾病来说,盐酸小檗碱的传统剂型还无法达到理想的治疗效果[3]。
纳米乳(NE)是由油相、水相、表面活性剂、助表面活性剂组成的脂质纳米给药载体。粒径范围为1~100 nm,液滴呈大小均匀的球形,外观呈透明或半透明。纳米乳是一种热力学稳定的胶体分散体系,制备工艺简单,所需能量低甚至能够自发形成。纳米乳处方中油相和表面活性剂、助表面活性剂等组成成分的增溶效果,可以显著提升难溶性药物的溶解度。纳米乳小且均匀的粒径,可透过胃肠道的水化层,改善胃肠上皮细胞对药物的吸收。纳米给药体系具有较低的表面张力,可提高膜的通透性,促进药物吸收,提高其生物利用度[4]。纳米乳自身的独特优势,使其具有广泛的临床应用范围,可通过口服、透皮、注射等多种途径给药[5-6]。
本研究结合纳米乳剂型的优势,制备了盐酸小檗碱口服纳米乳(BBR-NE),采用伪三元相图法和星点设计-效应面法优化了制备工艺,并对其体外特性进行表征,从而解决盐酸小檗碱口服给药剂型溶解度差、生物利用度低等问题,提高药物的疗效,为小檗碱的临床应用提供新的给药剂型[4]。
-
101A-2型干燥箱(上海实验仪器总厂);AG285十万分之一电子分析天平(瑞士Mettler Toledo公司);SB100D超声波清洗器(宁波新芝生物科技股份有限公司);Agilent 1100高效液相色谱仪(美国安捷伦科技有限公司);EPPENDORF 5804R 高速冷冻离心机(德国Eppendorf有限公司);DF-101S 集热式恒温加热磁力搅拌器(巩义市英峪予华仪器厂);Zetasizer Nano ZS90 测定仪(英国马尔文公司)。
-
盐酸小檗碱原料药(批号XC20170113,西安小草植物科技有限公司);盐酸小檗碱对照品(批号110713-201212,纯度>86.7 %,中国食品药品检定研究院);1,2-丙二醇(批号20181008,上海凌峰化学试剂有限公司);吐温-80(批号2015021,上海凌峰化学试剂有限公司);甘油(批号20191110,上海源叶生物科技有限公司);聚乙二醇400(PEG400,批号970248,上海浦东高南化工厂);聚氧乙烯氢化蓖麻油(RH-40,批号19523275L0,德国 BASF 公司);烷基糖苷0810(APG,平均聚合度1.2~1.8,批号170502,上海发凯化工有限公司);丙二醇单辛酸酯(Capryol 90,批号170857,上海嘉法狮贸易有限公司);肉豆蔻酸异丙酯(IPM,批号F20160731,国药集团化学试剂有限公司);橄榄油(批号F20180725,中国医药集团上海化学试剂公司);中链脂肪酸(MCT,批号193717,嘉法狮(上海)贸易有限公司);大豆油(批号20190801,江西金海棠药用油有限公司);蓖麻油(批号20180815,上海凌峰化学试剂有限公司);甲醇、乙腈(色谱纯,美国 TEDIA 有限公司);羟丙甲基纤维素(HPMC,批号H08827,阿拉丁生化科技股份有限公司);水为重蒸水。
-
色谱柱:Hypersil BDS C18柱(4.6 mm×250 mm,5 μm),流动相:乙腈-0.05 mol/L与磷酸二氢钾(0.5 %三乙胺,磷酸调至pH=3)等度洗脱,比例:30∶70,流速1.0 ml/min,紫外检测波长345 nm,柱温25 ℃,进样量20 μl。
-
精密称取盐酸小檗碱对照品1.0 mg,用甲醇稀释定容于10 ml量瓶中作为储备液,用甲醇逐级稀释成系列对照品溶液,按上述色谱条件进样测定。称取适量盐酸小檗碱纳米乳于10 ml 量瓶中,加入甲醇超声破乳30 min,10000 r/min离心5 min,取1 ml上清液于10 ml 量瓶中,加甲醇定容后过0.45 μm微孔滤膜即得供试品溶液。方法学考察表明,盐酸小檗碱在1.00~100.00 μg/ml浓度范围内呈良好的线性关系,回归方程为:Y=37.059 X+16.952 (r=0.999 9)。低、中、高浓度的盐酸小檗碱对照品溶液的日内精密度分别为0.68 %、1.64 %、0.80 %,日间精密度分别为0.95 %、0.23 %、013 %,加样回收率RSD分别为1.42 %、0.70 %、1.57 %,表明该方法可用于纳米乳中盐酸小檗碱的含量测定。
-
采用水滴加法制备盐酸小檗碱纳米乳。根据各类溶剂口服的安全性,筛选出以下各相备选溶剂,以BBR的溶解度为指标,确定各相最佳选择。
油相:Capryol 90、蓖麻油、橄榄油、大豆油、IPM、MCT;表面活性剂:吐温-80、APG、RH-40;助表面活性剂:1,2-丙二醇、PEG400、甘油。在具塞玻璃离心管中加入5 ml 上述各溶剂与过量盐酸小檗碱,置于恒温(25 ℃)振荡器振摇(100 r/min)72 h,得到以上各溶剂的饱和盐酸小檗碱溶液。取该饱和溶液于5000 r/min 条件下离心10 min,吸取1 ml上清液置于5 ml量瓶中,加甲醇稀释定容之后过0.45 μm微孔滤膜,按“2.1.1”项下色谱条件进样检测。
结果如图1所示:油相中盐酸小檗碱的溶解度从大到小依次为:Capryol 90> MCT>蓖麻油>大豆油>橄榄油>IPM;表面活性剂的顺序为:吐温-80>APG>RH-40;助表面活性剂的顺序为:1,2-丙二醇>甘油>PEG400。即盐酸小檗碱在Capryol 90、吐温-80、1,2-丙二醇中溶解度最大,故选择Capryol 90为油相,吐温-80为表面活性剂,1,2-丙二醇为助表面活性剂。
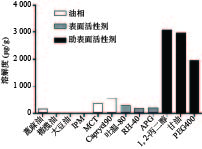
图 1 盐酸小檗碱在各相溶剂中的溶解度
-
按照上述筛选的结果,将表面活性剂和助表面活性剂按照Km分别为1∶1、1∶2、1∶3、2∶1、3∶1的比例混合,再取油相与此混合表面活性剂按照1∶9、2∶8;3∶7、4∶6、5∶5、6∶4、7∶3、8∶2、9∶1的质量比配制成混合溶液,在磁力搅拌下滴加蒸馏水。观察溶液的颜色、乳光和澄明度,判断临界点,记录体系由浑浊变澄清时的加水量,计算临界点时各组分的比例。用Origin 10.0软件绘制伪三元相图,图中各临界点连线下方的区域即为纳米乳区,比较该区域面积大小,确定表面活性剂与助表面活性剂的质量比[9-11]。结果见图2。图中阴影部分即为成乳区,且当Km为2∶1时达到最大,故将Km定为2∶1。然而在实际制备纳米乳过程中,处方中各组分的不同比例会产生交互影响作用,因此,需要进一步采用星点设计-效应面法优化盐酸小檗碱纳米乳的制备工艺。
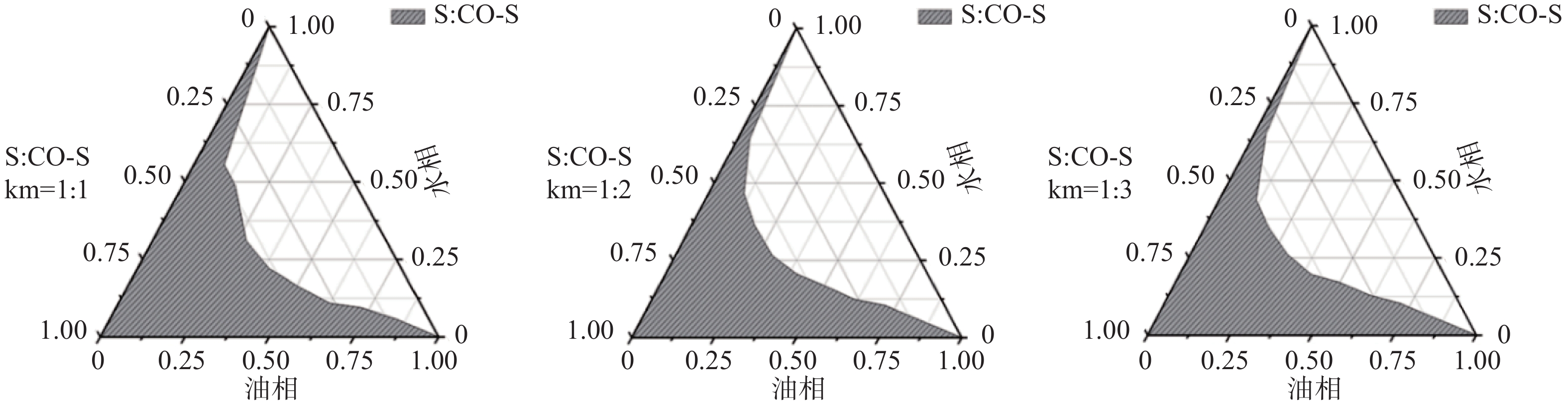
图 2 不同Km值BBR纳米乳的伪三元相图
-
选择对纳米乳影响较大的表面活性剂与助表面活性剂质量比Km(X1)和油相质量百分数(X2)作为考察因素,以多分散系数(PDI,Y1)和载药量(Y2)为考察指标。实验过程中发现当Km≥4时,大部分处方难以成乳;当Km≤1:2时,平均粒径>100 nm,故将Km范围定在1∶1至3∶1。当含油量增至0.5 g(50 %)时,平均粒径已接近100 nm,而当含油量范围在0.2~0.5 g(20 %~50 %)时,可将粒径控制在100 nm之内。根据单因素试验结果确定了Km的取值范围和油相质量百分数,并将两个评价指标进行归一化,将每个指标换算成0~1之间的“归一值(OD)”,并求算几何平均数,得总评“归一值”。计算公式如下: OD=(d1d2d3…... dk)1/k(k为指标数)。对取值越小越好的因素(PDI)和取值越大越好的因素(载药量)采用Hassan法[9]分别进行数学转换,求归一值dmin和dmax,公式如下:dmin=(Ymax−Yi)/(Ymax−Ymin);dmax=(Yi−Ymin)/(Ymax−Ymin)。采用2因素5水平星点设计优化处方,因素水平见表1,星点设计各组实验结果见表2。
表 1 星点设计因素水平表
因素 水平 −1.41 −1 0 +1 +1.41 X1(Km) 1∶1 1.29∶1 2∶1 2.71∶1 3∶1 X2(含油量/g) 0.2 0.24 0.35 0.46 0.5 表 2 星点设计各组实验结果
编号 X1 X2 平均粒径
(l
/nm)Y1 Y2
(mg/g)dmin dmax OD 1 0 −1.41 46.32 0.29 0.72 0.54 0 0 2 0 0 74.06 0.23 0.81 0.85 0.367 0.558 3 −1 1 85.3 0.25 0.91 0.78 0.820 0.800 4 0 0 74.06 0.23 0.84 0.85 0.53 0.67 5 1 −1 76.58 0.35 0.72 0.21 0.01 0.04 6 1.41 0 59.8 0.36 0.76 0.12 0.15 0.13 7 1 1 101.4 0.38 0.78 0.04 0.34 0.12 8 −1.41 0 58.68 0.21 0.80 1.00 0.36 0.60 9 −1 −1 45.8 0.21 0.84 0.99 0.51 0.71 10 0 0 74.06 0.23 0.81 0.85 0.37 0.56 11 0 0 74.06 0.23 0.81 0.85 0.37 0.56 12 0 0 74.06 0.23 0.80 0.85 0.37 0.56 13 0 1.41 91.88 0.39 0.95 0.00 1.00 0.00 对X1进行二次式回归,得到方程R1=0.23+0.06 A+3.22×10−3B−2.75×10−3AB+0.03A2+0.03B2 ,(r=0.970 8),P<0.001;对X2进行二项式回归,得到方程R2=0.81−0.04A+0.06B+1.37×10−3AB−0.01A2+0.01B2,(r=0.913 5),P<0.05;对总归一值(OD)进行二项式回归,方程为OD=0.87−0.28A+0.03B−0.01AB−0.03A2−0.34B2,(r=0.910 2),P<0.05。以OD作为指标时,为使方程有效,对其进行方差分析,结果见表3。由表3可知,模型的P < 0.05,说明OD与X1、X2回归方程的非线性关系显著。回归方程的相关系数r=0.910 2,表明模型能说明91.02 %响应值的变化,拟合情况良好,回归方程具有较好的代表性,能准确预测实际情况[12]。
表 3 星点设计-效应面法优化纳米乳处方方差分析表
来源 平方和 自由度 均方 F P 模型 1.48 5 0.18 5.88 0.0190 A-A 0.65 1 0.51 12.82 0.0090 B-B 7.73 1 3.600 0.15 0.7067 AB 4.40×10−5 1 0.016 8.74×10−3 0.9281 A2 6.32×10−3 1 0.37 0.13 0.7335 B2 0.83 7 0.026 16.41 0.0049 残差 0.35 3 0.058 纯误差 0.011 4 8.70×10-3 42.60 0.0017 总和 1.83 12 r 0.90 校正后r 0.82 C.V.% 35.29 信噪比 7.05 由拟合曲线可绘制X1、X2和OD值的关系,所绘制的三维效应面和二维等高线如图3所示。利用Design Expert 8.0软件的预测分析功能,根据拟合回归方程、三维效应面图和二维等高线图综合分析,得到最佳处方为X1= 0,X2=0.53,预测值OD为0.6214。故优化后的盐酸小檗碱纳米乳处方为:油相Capryol 90占体系的32.84 %,表面活性剂吐温-80占体系的33.90 %,助表面活性剂1,2-丙二醇占体系16.95 %,水相占体系15.25 %[4]。
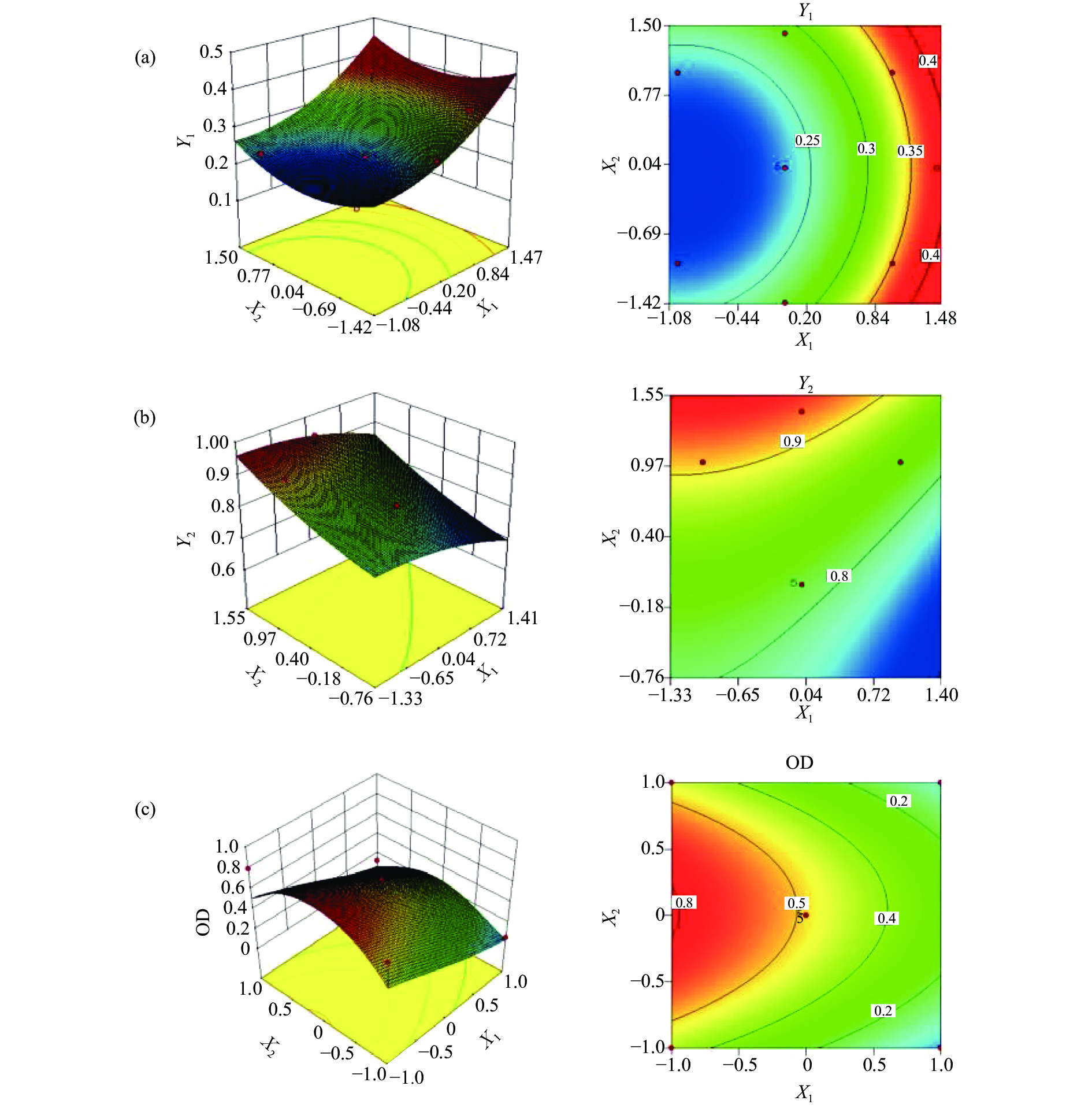
图 3 因变量Y随自变量X变化的三维效应面和二维等高线图
-
对最优处方进行处方验证,结果如表4所示,预测值OD为0.6214,实际OD值为0.6510,偏差为4.76 %。表明预测值与实测值之间偏差较小,表明星点设计-效应面法(CCD-RSM)可用于筛选盐酸小檗碱纳米乳,筛选的处方预测良好、可靠。
表 4 最优处方验证结果
评价指标 实测值 实际OD值 预测OD值 偏差(%) PDI 0.235±0.03 0.6510 0.6214 4.76 载药量(mg/g) 0.829 -
取适量BBR-NE于10 ml量瓶中,加甲醇超声破乳30 min,加甲醇稀释定容。4 ℃离心(10000 r/min,5 min),吸取1 ml上清液置于10 ml容量瓶中,加甲醇稀释定容后即得样品溶液。按“2.1.1”项下色谱条件进样检测,计算纳米乳的载药量。测定结果如表5。
表 5 盐酸小檗碱纳米乳的载药量测定结果(
$\bar x $ ±s ,n=3)批号 盐酸小檗碱(mg/g) RSD(%) 201118 0.83±0.01 0.16 201119 0.83±0.02 0.21 201120 0.83±0.01 0.17 -
按照最优处方制备BBR-NE,稀释到适宜浓度,均匀分散后,采用马尔文激光粒度分析仪测定纳米乳的粒径大小。粒径结果如图4和图5所示。所制备的BBR-NE粒径分布范围窄且呈正态分布,平均粒径为(68.85±8)nm,PDI为(0.245±0.03),表明该制剂粒径分布及均匀性均符合纳米乳制剂要求。
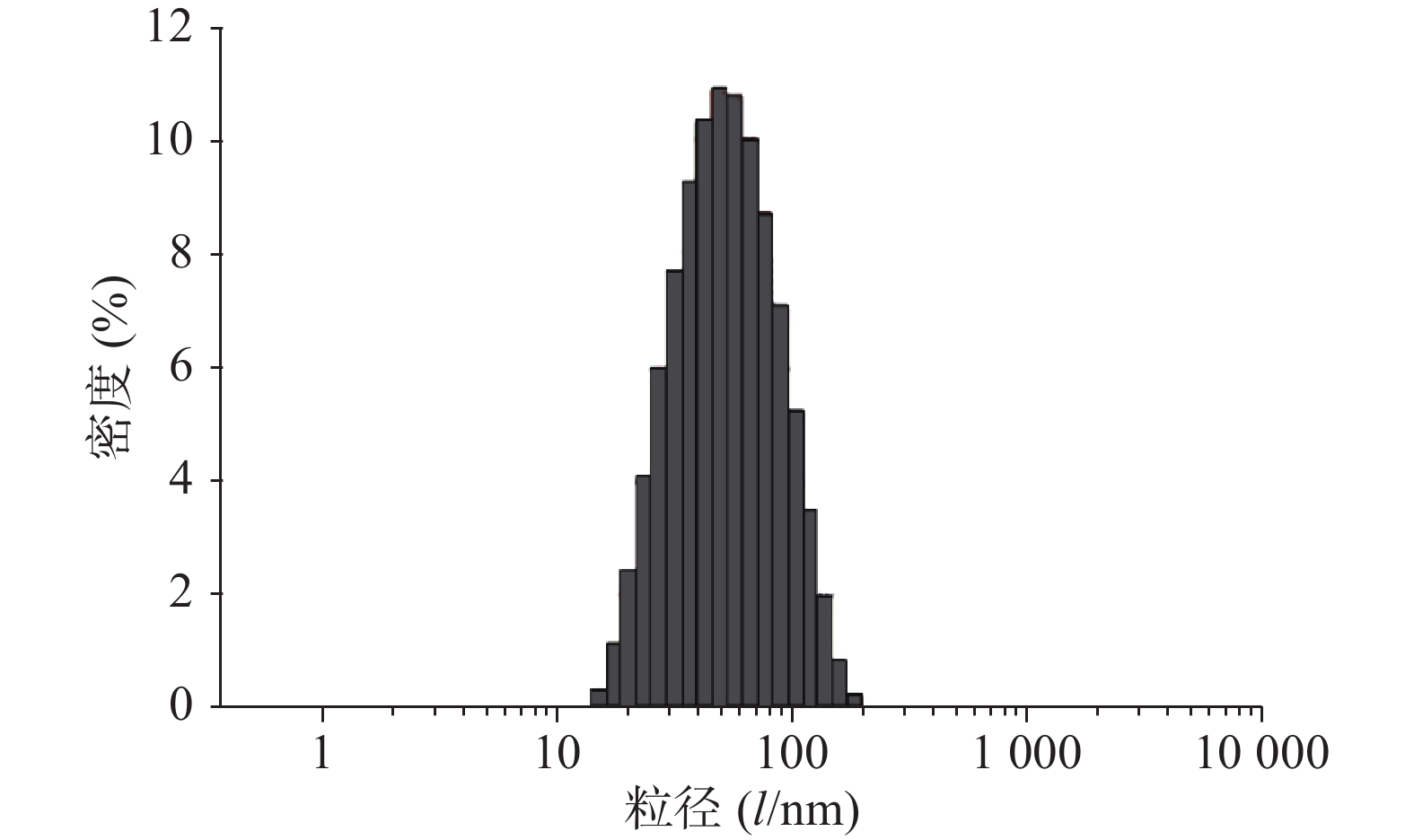
图 4 最优处方(201119)纳米乳粒径分布图
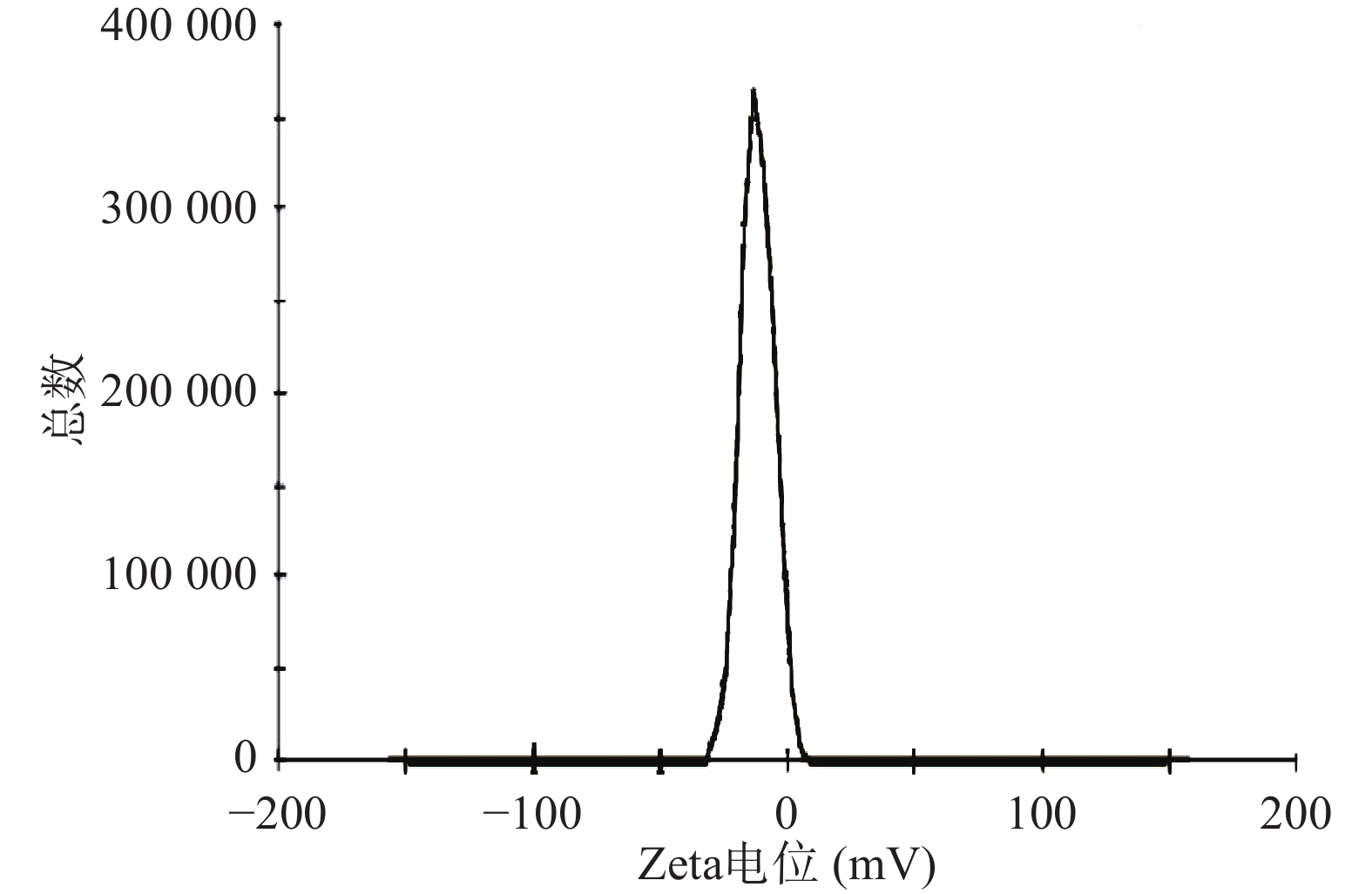
图 5 最优处方(201119)纳米乳电位分布图
-
采用透射电镜(TEM)观察所制备的BBR-NE的形态。取适量BBR-NE滴于铜网上,自然干燥后滴加2 %磷钨酸(pH=7.4)溶液于铜网上负染3 min,晾干后置于透射电镜下观察其外观形貌。最优处方制备的纳米乳的透射电镜如图6所示。结果表明,BBR-NE呈均一圆整的球形,具明显层状结构,粒径大小约为68 nm。

图 6 最优处方(201119)纳米乳的透射电镜图
-
取制备好的盐酸小檗碱纳米乳2 ml,置于透析袋(分子截留值为7 000)中。于pH 1.2的人工胃液中释放2 h,再于pH 6.8的人工小肠液中释放22 h,溶出介质体积满足漏槽条件。将透析袋置于离心管中(含30 ml透析液),在恒温振荡器(37 ℃,100 r/min)中释放24 h,分别于0.5、1、2、3、4、6、8、10、12、16、20、24 h定时取样1 ml,随后立即补加等温等体积释放介质。收集的样品过0.45 μm滤膜后,按照“2.1.1”项下色谱条件进样测定,记录峰面积,计算累积释放度。释放曲线如图7所示,拟合方程如表6所示。
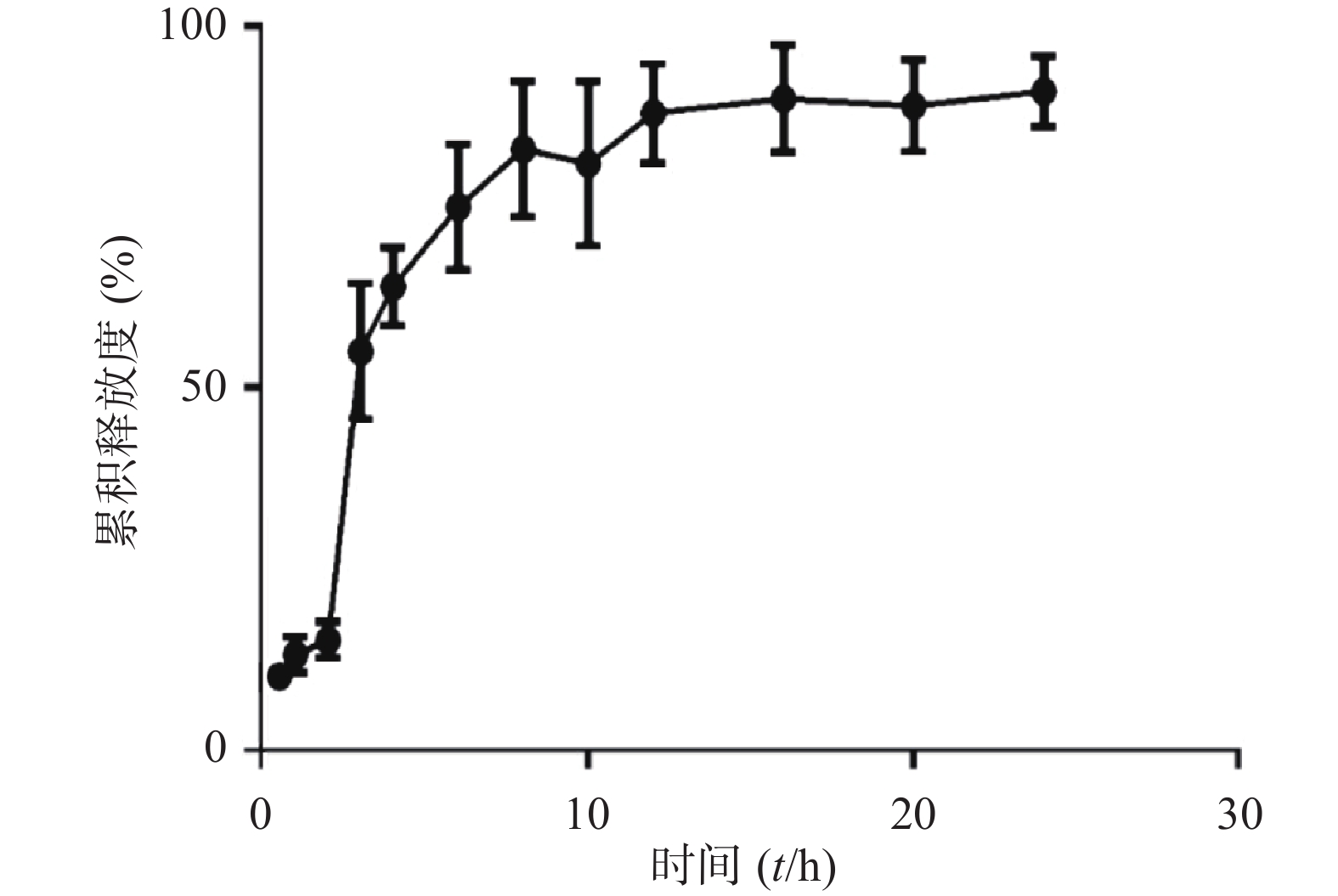
图 7 盐酸小檗碱纳米乳的体外释放曲线 (
$\bar x $ ±s ,n=6)表 6 盐酸小檗碱纳米乳释药曲线的拟合方程
释药模型 拟合方程 r 零级动力学方程 Q=29.352 t–4.123 0.816 4 一级动力学方程 ln(1–Q)=92.395 t+0.240 0.962 3 希古契(Higuchi)方程 Q=20.952 t 1/2–6.735 0.889 6 由释放曲线可以看出,BBR-NE在人工胃液中(0~2 h)累积释放度很低(<20%),进入人工小肠液后(2~24 h)释放度逐渐上升,累积释放度达到80 %以上,表明该制剂具有一定缓释效果,其释药行为符合一级动力学方程。
-
纳米乳的处方筛选过程中,目前常采用绘制伪三元相图、正交试验设计法、星点设计-效应面法等方法。其中,伪三元相图法的精度不高,常用于初筛[13-14];正交试验设计法在因素水平较少时试验次数虽不多,但由于采用线性模型拟合,预测性较差[15];而星点设计-效应面法各因素对效应的影响拟合结果多数是非线性,可从效应面上选择最佳效应区域,适合于多因素、多水平的处方优化。
在应用伪三元相图筛选Km时,随着表面活性剂和助表面活性剂质量比的增加,形成纳米乳区域的面积逐渐增加,当Km为2∶1 时最大,而后稍有减小。这是由于加入吐温-80的比例增加,纳米乳的乳化作用增强。因此需要综合考虑选择最佳的Km值。
本研究在单因素筛选的基础上,利用星点设计-效应面法,以载药量及外观形态为考察指标,优化并确定了盐酸小檗碱纳米乳的最优处方:油相 Capryol 90占体系32.84 %,表面活性剂吐温−80占体系33.90 %,助表面活性剂1,2-丙二醇占体系16.95 %,水相占体系15.25 %,载药量为0.829 mg/g。 外观形态分析,表明所制备的BBR-NE 呈圆整均一的球体且具有明显的层状结构,平均粒径为 (68.85±8) nm,呈正态分布,多分散系数为(0.245±0.03),说明该制剂粒径大小符合纳米乳粒径要求且均匀性好[4]。由验证试验结果可知,该制备工艺稳定可行,有望为盐酸小檗碱临床应用提供一种新的给药剂型。
Preparation and in vitro evaluation of berberine hydrochloride nanoemulsion oral drug delivery systems
-
摘要:
目的 制备盐酸小檗碱纳米乳,优选其处方组成和制备工艺,并对其外观、粒径及体外释放行为等进行评价。 方法 采用水滴加法制备盐酸小檗碱纳米乳,绘制纳米乳伪三元相图。选择星点设计-效应面法对纳米乳处方进行优化,确定最优处方组成比例。对制备的盐酸小檗碱纳米乳的粒径、电位、外观形态、体外释药行为等进行表征。 结果 确定了盐酸小檗碱纳米乳的最优处方为油相Capryol 90占体系的32.84 %,表面活性剂吐温-80占体系的33.90 %,助表面活性剂1,2-丙二醇占体系16.95 %,水相占体系15.25 %,制备的纳米乳呈澄清透明、形状规则、大小均一的球状,平均粒径为(68.85±8)nm,多分散系数为(0.245±0.03),载药量为0.83 mg/g。纳米乳的体外释药行为考察,表明其具有一定的缓释效果,其体外释药行为符合一级释放动力学方程。 结论 本研究制备的盐酸小檗碱纳米乳可为该药的临床用药提供新的给药剂型。 Abstract:Objective To prepare berberine hydrochloride nanoemulsion, optimize its formulation composition and preparation process, and investigate its in vitro characteristics. Methods BBR-NE was prepared by water drop addition and pseudo-ternary phase diagram was drawn. The formulation of NE was optimized by central composite design-response surface methodology to choose the optimal formulation composition. The particle size, potential and appearance of the prepared BBR-NE were characterized. Results The optimal prescription of BBR-NE was determined as the oil phase Capryol 90 accounted for 32.84% of the system, the surfactant Tween-80 accounted for 33.90%, the co-surfactant 1,2-propylene glycol accounted for 16.95%, and water relative system accounted for 15.25%. The prepared NE was clear and transparent in appearance, regular in shape and uniform in size, with an average particle diameter of (68.85±8) nm, polydiseperse index of (0.245±0.03) and drug loading of 0.83 mg/g. The in vitro drug release results of NE showed that the in vitro drug release behavior was passive diffusion, which had a certain slow releasing effect and met the first-order release equation. Conclusion The BBR-NE can provide a new dosage form for the clinical use of berberine. -
刺梨,又名文先果,为蔷薇科植物单瓣缫丝花(Rosa roxburghii Tratt.)的果实,主产于贵州、云南、广西等省份。具有健脾消食,收敛止泻之功[1]。刺梨含有碳水化合物、维生素[2]、氨基酸[3]、脂肪酸[4]、微量元素[5]等丰富的营养物质,以及委陵菜酸、刺梨酸、刺梨苷、野蔷薇苷[6]、野鸦春酸、刺梨素、槲皮素等黄酮类[7]、三萜类、有机酸类、多酚类[8]、多糖类[9]等活性物质[10]。具有抗氧化[11]、抗菌[12]、降血糖[13]、降血脂[14]、解酒护肝[15]、保护与修复胃溃疡黏膜[16]、抗衰老[17]、抗辐射[18]、抗缺氧[19]、调节机体免疫功能[20]、抗肿瘤[21-22]等作用,食用[23-24]及药用[25]价值极高,目前食品工业及保健品行业应用广泛,药用以民间及民族药应用较广,部分中成药中也有应用。现行标准中刺梨作为药用仅在少数省份中药饮片炮制规范中出现,标准内容基本局限在来源鉴定、性状鉴定上,缺乏质量控制的指标性限定。本实验在新修安徽省中药饮片炮制规范及中药材标准项目的基础上,以现行版《中国药典》(四部)指导原则为依据开展。除特征图谱标准外,显微鉴定、薄层鉴定、常规检查等也作为标准制定的考察内容。
1. 仪器与材料
1.1 仪器
分析天平(METTLER TOLEDO公司);赛默飞U3000系列液相色谱仪(赛默飞世尔科技有限公司);赛默飞Vanquish Core系列液相色谱仪(赛默飞世尔科技有限公司);色谱柱:赛默飞AcclaimTM120 C18(4.6 mm×250 mm ,5 μm,120Å)、安捷伦ZORBAX SB-C18(4.6 mm×250 mm,5 μm)、依利特C18(4.6 mm×250 mm,5 μm);薄层色谱自动点样仪及成像系统(瑞士CAMAG公司)。
1.2 试药
共收集8批次刺梨药材(按CL-01-00X顺序编号),经海军军医大学药学系生药学教研室黄宝康教授鉴定为蔷薇科植物单瓣缫丝花(Rosa roxburghii Tratt.)的果实。刺梨果对照药材(中国食品药品检定研究院,批号:121356-200401,纯度:供薄层鉴别用);异槲皮苷对照品(中国食品药品检定研究院,批号:111809-201804,纯度:92.9%);色谱级乙腈(国药集团)。
2. 方法与结果
2.1 特征图谱
2.1.1 色谱条件与系统适用性试验
以十八烷基硅烷键合硅胶为填充剂;乙腈为流动相A,0.1%甲酸水溶液为流动相B,按表1中的规定进行梯度洗脱,流速为0.8 ml/min,柱温25 ℃,检测波长为360 nm。理论板数按异槲皮苷峰计算应不低于3 000。
表 1 流动相的组成时间(t/min) 流速(ml/min) A(%) B(%) 0 0.8 8 92 10 0.8 10 90 20 0.8 12 88 40 0.8 14 86 50 0.8 16 84 60 0.8 18 82 63 0.8 12 88 66 0.8 8 92 2.1.2 参照物溶液的制备
取刺梨对照药材l g,加入80%甲醇溶液50 ml,加热回流2 h,取出,放冷,滤过,滤液浓缩至干,加水30 ml使溶解,用饱和的正丁醇萃取4次,每次30 ml,合并正丁醇溶液,蒸干,加甲醇定容至25 ml量瓶中,作为对照药材参照物溶液。另取异槲皮苷对照品加甲醇溶解,制成每1 ml含40 μg 的溶液,作为对照品参照物溶液。再精密量取对照药材参照物溶液2 ml,对照品参照物溶液1 ml,混匀,得混合参照物溶液,微孔滤膜过滤,即得。
2.1.3 供试品溶液的制备
取刺梨粉末约2 g,加入80%甲醇溶液50 ml,加热回流2 h,取出,放冷,滤过,滤液浓缩至干,加水30 ml使溶解,用饱和的正丁醇萃取4次,每次30 ml,合并正丁醇溶液,蒸干,加甲醇定容至25 ml量瓶中,再精密量取2 ml及对照品参照物溶液1 ml,混匀制成供试品溶液,即得。
2.1.4 测定方法
分别精密吸取上述参照物溶液与供试品溶液各20 μl,注入液相色谱仪,记录色谱图,即得(图1~图4)。
2.1.5 线性范围考察
精密称取纯度为97.2%的异槲皮苷对照品适量置50 ml容量瓶中,加甲醇稀释至刻度,摇匀,得对照品溶液储备液,分别精密量取一定量的对照品储备液稀释成浓度分别为0.0371、0.0743、0.1114、0.1485、0.1857、0.2228、0.3710 mg/ml的对照品溶液,注入液相色谱仪,测定含量,以浓度为纵坐标、峰面积为横坐标得标准曲线Y=905.02X−0.360 9 (r=0.999 7)。
2.1.6 精密度及稳定性试验
精密量取混合参照物溶液20 μl注入液相色谱仪,测定加入异槲皮苷对照品峰面积,平行测定5次,计算保留时间及峰面积RSD,分别为0.1%、0.1%。精密量取混合参照物溶液20 μl注入液相色谱仪,分别在0、2、4、8、16、24 h测定加入异槲皮苷对照品峰面积,计算保留时间和峰面积RSD,分别为0.3%、1.0%。
2.1.7 样品测定
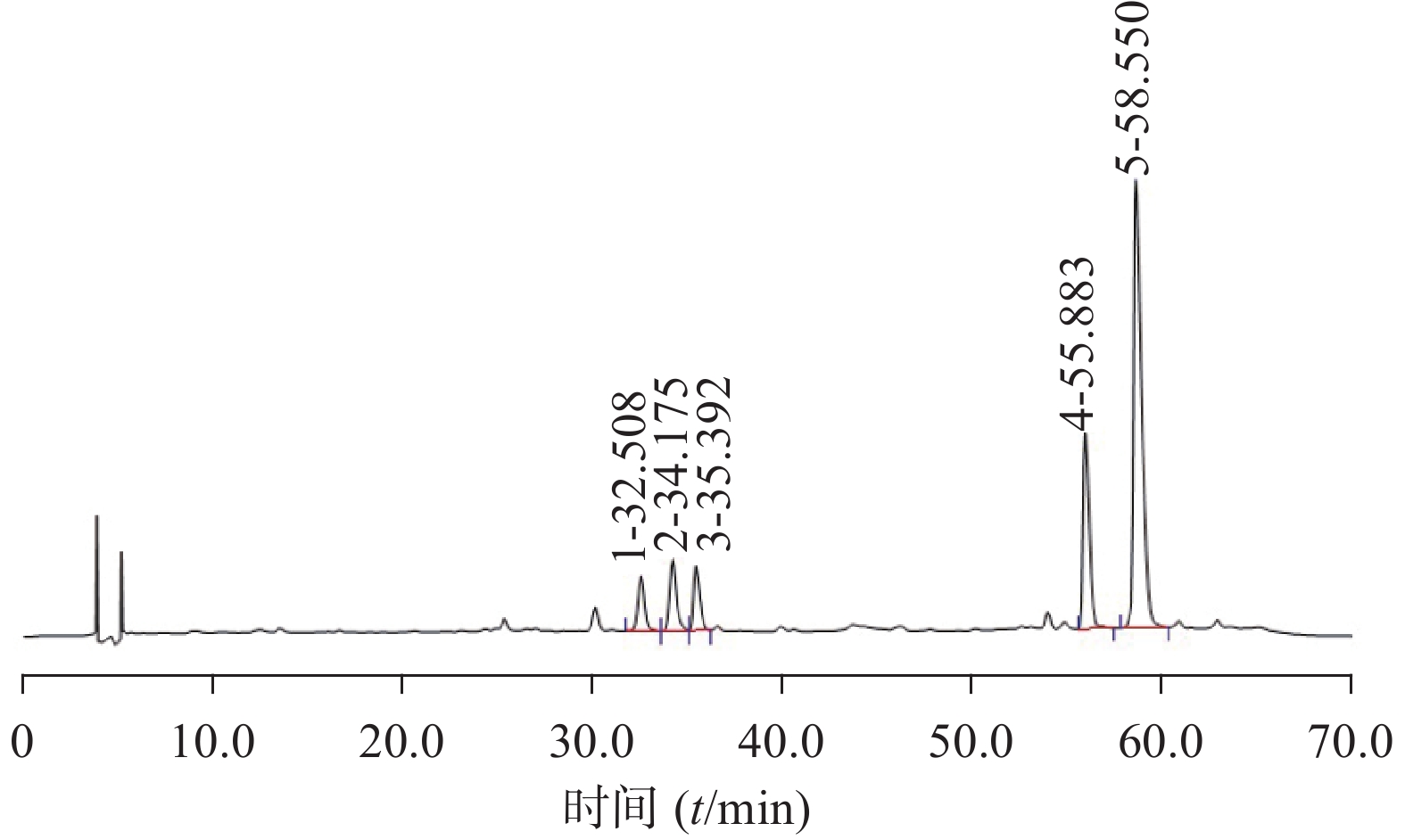
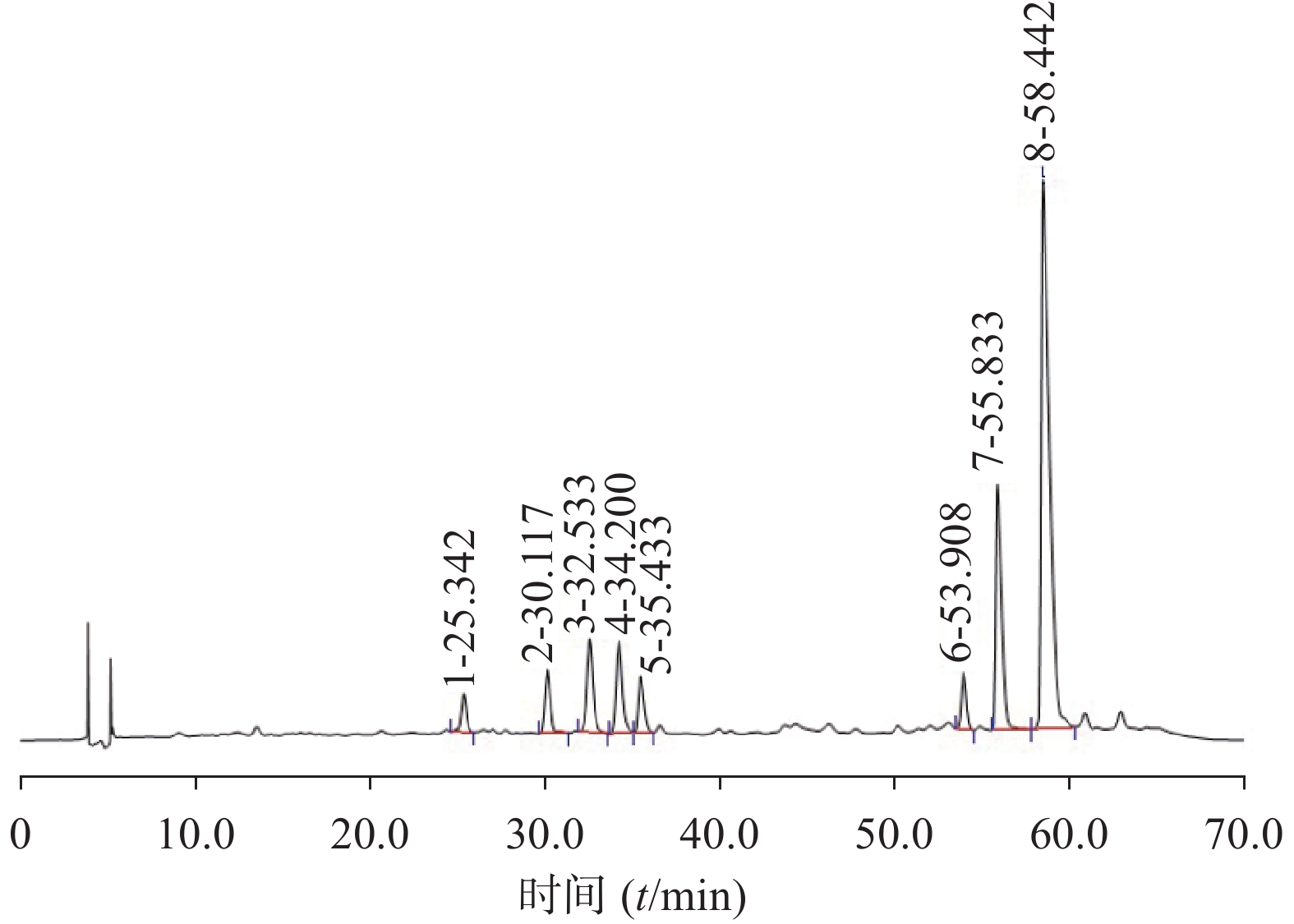
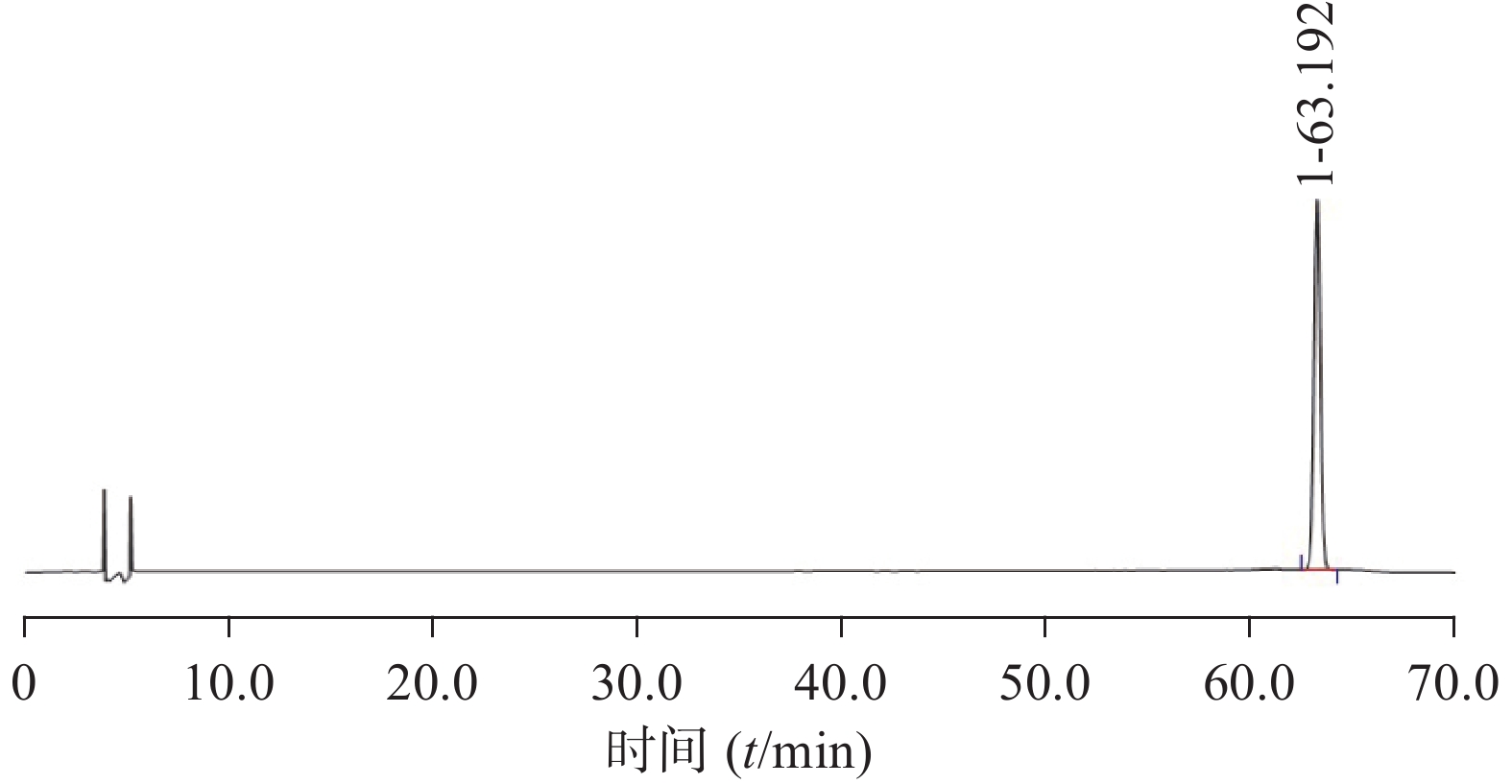
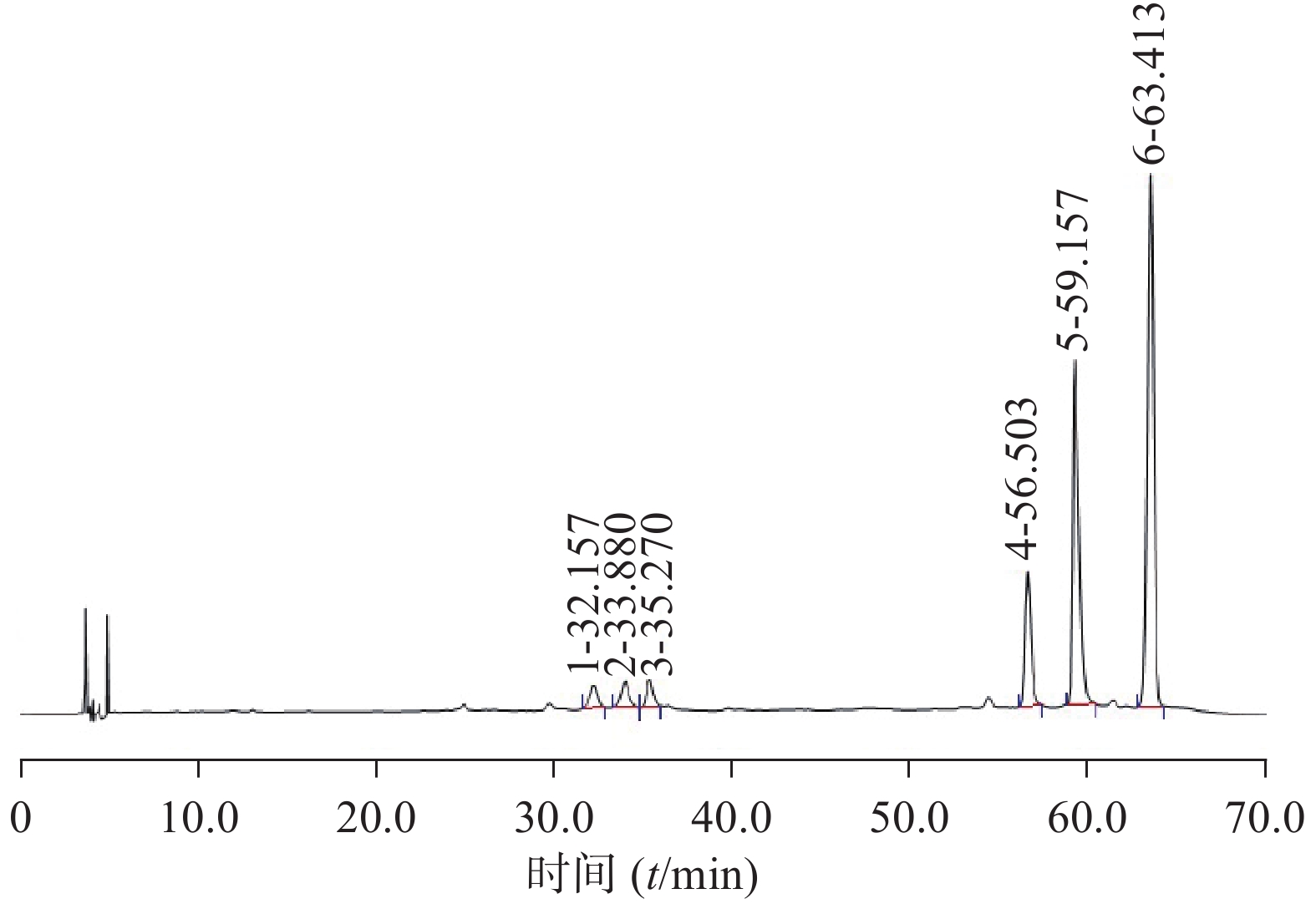
测定8批样品,记录加入对照品及特征峰的相对保留时间,计算相对保留时间偏差,结果发现对照药材及样品共有5个峰,加入异槲皮苷对照品后,显示6个特征色谱峰(表2、表3),根据保留时间,选择相对保留时间为0.52、0.54、0.56、0.89、0.93时的色谱峰为特征色谱峰,可控制偏差范围±5%。参考《中国药典》(2020年版)四部通则指导原则的有关规定,拟定:供试品色谱中应呈现6个特征峰,并应与对照药材参照物色谱峰中的5 个特征峰保留时间相对应,另有1峰应与加入的异槲皮苷对照品参照物峰保留时间相对应(图5)。
表 2 对照药材特征峰相对保留时间序号 峰 保留时间(t/min) 相对保留时间 1 峰1 32.419 0.52 2 峰2 34.077 0.54 3 峰3 35.339 0.56 4 峰4 55.781 0.89 5 峰5 58.434 0.93 6 异槲皮苷 62.711 1.00 表 3 样品共有峰相对保留时间(n=8)样品 峰1 峰2 峰3 峰4 峰5 异槲皮苷峰 对照药材 0.52 0.55 0.57 0.89 0.93 1.00 CL-01-001 0.52 0.54 0.57 0.89 0.93 1.00 CL-01-002 0.52 0.54 0.56 0.89 0.93 1.00 CL-01-003 0.52 0.55 0.57 0.89 0.93 1.00 CL-01-004 0.52 0.55 0.57 0.89 0.93 1.00 CL-01-005 0.52 0.54 0.56 0.89 0.93 1.00 CL-01-006 0.52 0.54 0.56 0.89 0.93 1.00 CL-01-007 0.51 0.54 0.56 0.89 0.93 1.00 CL-01-008 0.51 0.54 0.56 0.89 0.93 1.00 平均值 0.52 0.54 0.56 0.89 0.93 1.00 RSD(%) 0.8 0.9 0.9 0.0 0.0 0.0 2.1.8 不同色谱条件对相对保留时间的影响
分别采用赛默飞AcclaimTM120 C18(5 μm,4.6 mm×250 mm)、安捷伦ZORBAX SB-C18(5 μm, 4.6 mm×250 mm)、依利特C18(5 μm ,4.6 mm×250 mm)色谱柱考察色谱柱的影响;柱温在20、25、30 ℃考察柱温的影响;流速为0.8、1.0、1.2 ml/min考察流速的影响;以及不同实验室考察检测环境变化的影响。根据测定数据分析,色谱柱、柱温、流速、不同实验室(表4)等,对特征峰相对保留时间具有一定的影响,其中,对峰1、峰2、峰3影响较大,但各色谱峰的影响RSD小于5%,属于可接受范围。
表 4 不同色谱条件对保留时间的影响(n=3)色谱条件 峰1 峰2 峰3 峰4 峰5 异槲皮苷 色谱柱 赛默飞 0.52 0.55 0.57 0.89 0.93 1.00 依利特 0.50 0.53 0.54 0.88 0.92 1.00 安捷伦 0.49 0.50 0.53 0.88 0.93 1.00 平均値 0.50 0.53 0.55 0.88 0.93 1.00 RSD(%) 2.5 3.9 3.1 0.5 0.5 0.0 柱温(t/ ℃) 20 0.53 0.56 0.58 0.89 0.94 1.00 25 0.52 0.55 0.57 0.89 0.93 1.00 30 0.49 0.52 0.54 0.89 0.92 1.00 平均値 0.51 0.54 0.56 0.89 0.93 1.00 RSD(%) 3.3 3.1 3.0 0.0 0.9 0.0 流速(ml/min) 0.8 0.52 0.55 0.57 0.89 0.93 1.00 1.0 0.48 0.51 0.53 0.88 0.92 1.00 1.2 0.46 0.49 0.52 0.89 0.91 1.00 平均値 0.49 0.52 0.54 0.89 0.92 1.00 RSD(%) 5.1 4.8 4.0 0.5 0.9 0.0 实验室 209室 0.52 0.55 0.57 0.89 0.93 1.00 703室 0.51 0.53 0.56 0.89 0.93 1.00 平均値 0.52 0.54 0.57 0.89 0.93 1.00 RSD(%) 1.0 1.8 1.9 0.0 0.0 0.0 2.2 鉴定
2.2.1 性状鉴定
刺梨以干燥果实入药,药材整体上呈扁球形。表面黄褐色,贮藏时间和干燥方式对色泽稍有影响,新鲜果实直接烘干颜色稍浅,贮藏较久颜色加深,密被针刺,针刺脱落处成点状凸起,有的并具褐色斑点;先端常有黄褐色宿存的花萼5瓣,亦披针刺。种子多数,着生于萼筒基部凸起的花托上,卵圆形,浅黄色,质硬,种仁乳白色。气微香,味酸、涩、微甜[26]。
2.2.2 显微鉴定
粉末呈棕色至棕褐色,石细胞较多,形状多样。非腺毛为单细胞毛,壁不甚厚。纤维管胞长梭形,成群。草酸钙簇晶偶见,角多钝。胚乳细胞成片,内含脂肪油滴。
2.2.3 薄层鉴定
薄层鉴别是中药鉴定常用的鉴别方法,多国药典采用,实验参考有关中成药制剂制定薄层鉴别方法[27],具体方法:取本品粉末1 g,加水50 ml,超声处理15 min,取上清液用盐酸饱和的乙醚振摇提取2次(每次20 ml),合并乙醚液,挥干,残渣加甲醇1 ml使溶解,作为供试品溶液。另取刺梨果对照药材1 g,同法制成对照药材溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各10 μl分别点于同一硅胶G薄层板上,以甲苯(水饱和)-醋酸乙酯-甲酸(6:3:1 )为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。
2.3 检查
检查项依据《中国药典》通则分别测定了所有收集批次刺梨的水分及总灰分。水分测定采用通则0832第二法测定,8批次刺梨水分在10.02%~13.77%之间。以统计学方法分析数据,水分限度以
$\mu =\bar {\rm{x}} + ts/\sqrt {\rm{n}} + {\rm{MU}}$ ($\bar {\rm{x}} $ 是样本的平均数;t是置信水平为99%的t检测值(单尾);s是样本的标准偏差;n是样本的批数;MU是不确定度评估。以下公式注同)计算,结果为14.20%,故拟定水分不得过14.0%。总灰分采用通则2302所述方法测定,8批刺梨的总灰分在3.28%~3.51%之间。以统计学方法分析数据,总灰分限度以公式$\mu =\bar {\rm{x}} + ts/\sqrt {\rm{n}} + {\rm{MU}}$ 计算,由实验数据及统计计算得灰分的限度为3.78%,考虑到样本量以及前期实验测定的结果,拟定总灰分不得过5.0%。2.4 浸出物
照醇溶性浸出物测定法(《中国药典》2020年版四部 通则2201)项下的冷浸法测定,用稀乙醇作溶剂,共测定8批次刺梨浸出物,浸出物在21.6%~51.5%之间。以统计学方法分析数据,浸出物限度以公式
$\mu =\bar {\rm{x}} - ts/\sqrt {\rm{n}} -{\rm{MU}} $ ,结果为20.5%,故拟定浸出物不得少于20.0%。2.5 标准其他项目
刺梨味酸、涩、甘,归肝、脾经。具有健胃、消食,解暑之功。用于食积饱胀,肠炎腹泻等。用法为内服,生食或煎汤,3~5枚。贮藏于通风干燥处,防蛀[26]。
3. 讨论
中药质量标准的制定既需要来源鉴定、性状鉴定、检查项等常规的鉴别内容,还需要保证中药药效的物质限度,目前常用的质量控制方法包括有效成分的指纹图谱、特征图谱、一测多评[28]、质量标志物Q-Marker质量控制等。中药Q-Marker通过药效、功效成分、质量稳定与可测性等,全方位控制中药质量,但是Q-Marker质量控制对所含化学成分能够明确相关功效的物质要求较高[29],目前刺梨中具有专属性强、药效相对明确物质尚不明确,通过Q-Marker质量控制有一定难度。有效成分指纹图谱、一测多评等质量控制方法需要明确刺梨中的专属成分,刺梨酸、刺梨苷等是具有专属性强、结构明确、具有一定活性的物质,可用作质量控制的标志性成分,但符合中药标准物质条件的产品,权威部门尚不能提供,将会对药材的上市检测带来不便。刺梨中含有丰富的黄酮类成分包括槲皮素、山奈素等[10],具有较明确的药理活性,但是实验过程中槲皮素等成分液相色谱行为表现较差,检出率、重现性、稳定性以及提取方法的复杂程度等不适于特征图谱标准的制定,故选择结构类似,中国食品药品检定研究院可以足量供应的异槲皮苷作为内标对照品。实验过程中还考察了色谱柱、流速等对色谱峰的影响,色谱条件的变化对出峰数量、出峰顺序无影响,对相对保留时间有一定的影响,其中色谱柱和流速等对峰1、2、3影响稍大,但RSD小于5%,重现性、稳定性良好,属于可接受范围。
实验采用全波长检测,除分析360 nm黄酮类为主的成分特征峰之外,还考察了刺梨苷、野蔷薇苷为主的三萜皂类常用检测波长203 nm下的特征色谱峰行为,近末端吸收情况下溶剂干扰较大、色谱峰形较差、干扰峰较多[30],且特征化合物刺梨苷等对照品无法从法定机构获得,故未选择此条件下的特征图谱用于标准制定。针对中药功效成分复杂、结构不明确等特点,从整体上控制质量是保证药效的有效途径之一。薄层色谱法可充分利用展开后斑点信息[31],全面展示中药成分的差异,且操作简单、快速,对仪器要求不高,结合特征图谱等可以更全面地整体性控制中药的质量,因此,实验中增加了药材的薄层色谱鉴别。
4. 结论
实验采用对照药材建立对照特征图谱,通过加入结构明确的对照品标定特征对照峰并对特征成分进行说明,检出的特征峰数,包含成分不明确的色谱峰,在标准中要求供试品能够检出与对照药材相一致的色谱峰。同时参考相关文献规范了性状鉴别、增加了显微鉴别、薄层鉴别,对常规检查项也做了相应的检测,制定了限度标准。研究结果在安徽省中药饮片炮制规范及安徽省中药材标准新修项目的基础上完成,为相关地方标准的制定提供了实验依据。
-
表 2 星点设计各组实验结果
编号 X1 X2 平均粒径
(l
/nm)Y1 Y2
(mg/g)dmin dmax OD 1 0 −1.41 46.32 0.29 0.72 0.54 0 0 2 0 0 74.06 0.23 0.81 0.85 0.367 0.558 3 −1 1 85.3 0.25 0.91 0.78 0.820 0.800 4 0 0 74.06 0.23 0.84 0.85 0.53 0.67 5 1 −1 76.58 0.35 0.72 0.21 0.01 0.04 6 1.41 0 59.8 0.36 0.76 0.12 0.15 0.13 7 1 1 101.4 0.38 0.78 0.04 0.34 0.12 8 −1.41 0 58.68 0.21 0.80 1.00 0.36 0.60 9 −1 −1 45.8 0.21 0.84 0.99 0.51 0.71 10 0 0 74.06 0.23 0.81 0.85 0.37 0.56 11 0 0 74.06 0.23 0.81 0.85 0.37 0.56 12 0 0 74.06 0.23 0.80 0.85 0.37 0.56 13 0 1.41 91.88 0.39 0.95 0.00 1.00 0.00  下载: 导出CSV
下载: 导出CSV
表 3 星点设计-效应面法优化纳米乳处方方差分析表
来源 平方和 自由度 均方 F P 模型 1.48 5 0.18 5.88 0.0190 A-A 0.65 1 0.51 12.82 0.0090 B-B 7.73 1 3.600 0.15 0.7067 AB 4.40×10−5 1 0.016 8.74×10−3 0.9281 A2 6.32×10−3 1 0.37 0.13 0.7335 B2 0.83 7 0.026 16.41 0.0049 残差 0.35 3 0.058 纯误差 0.011 4 8.70×10-3 42.60 0.0017 总和 1.83 12 r 0.90 校正后r 0.82 C.V.% 35.29 信噪比 7.05
下载: 导出CSV
表 5 盐酸小檗碱纳米乳的载药量测定结果(
$\bar x $ ±s ,n=3)批号 盐酸小檗碱(mg/g) RSD(%) 201118 0.83±0.01 0.16 201119 0.83±0.02 0.21 201120 0.83±0.01 0.17
下载: 导出CSV
表 6 盐酸小檗碱纳米乳释药曲线的拟合方程
释药模型 拟合方程 r 零级动力学方程 Q=29.352 t–4.123 0.816 4 一级动力学方程 ln(1–Q)=92.395 t+0.240 0.962 3 希古契(Higuchi)方程 Q=20.952 t 1/2–6.735 0.889 6
下载: 导出CSV
-
[1] ZHANG J H, ZHANG J F, SONG J, et al. Effects of berberine on diabetes and cognitive impairment in an animal model: the mechanisms of action[J]. Am J Chin Med,2021,49(6):1399-1415. doi: 10.1142/S0192415X21500658 [2] SHEN Z Q, WANG J, TAN W F, et al. Berberine inhibits colorectal tumor growth by suppressing SHH secretion[J]. Acta Pharmacol Sin,2021,42(7):1190-1194. doi: 10.1038/s41401-020-00514-2 [3] 赵西子, 李文芳, 邢彦超, 等. 小檗碱体内药代动力学及药理活性研究进展[J]. 辽宁中医药大学学报, 2020, 22(10):86-90. [4] 王婷. 小檗碱纳米乳口服给药系统的构建及体内外评价[D]. 济南: 山东中医药大学, 2020. [5] 邹灵辉, 丁文雅, 黄秋艳, 等. 纳米乳在中药制剂领域的应用优势及其研究进展[J]. 中国实验方剂学杂志, 2021, 27(18):217-226. [6] 王敏娟, 李惠民, 冯锁民, 等. 纳米乳载药研究进展[J]. 化工科技, 2020, 28(1):69-75. [7] 唐晓萌, 骆锦前, 汪五清, 等. 白术黄连微丸结肠靶向胶囊的制备及体外释放研究[J]. 药学实践杂志, 2021, 39(1):29-34. doi: 10.12206/j.issn.1006-0111.202008109 [8] 唐晓萌. 基于两步释放的术连微丸口服结肠靶向胶囊的研制及体内外评价[D]. 上海: 海军军医大学, 2019. [9] 张琳琳, 李小芳, 谢龙, 等. 星点设计-效应面法优化基于甘草酸的葛根素纳米乳及其体外释放研究[J]. 中草药, 2020, 51(12):3180-3186. doi: 10.7501/j.issn.0253-2670.2020.12.009 [10] 赵惠茹, 郭秋芸, 蒋本进, 等. 星点设计-效应面法优化芸香苷纳米乳的制备工艺[J]. 中国药师, 2018, 21(2):204-208. doi: 10.3969/j.issn.1008-049X.2018.02.004 [11] WAKISAKA S, NISHIMURA T, GOHTANI S. O/W nano-emulsion formation using an isothermal low-energy emulsification method in a mixture of polyglycerol polyricinoleate and hexaglycerol monolaurate with glycerol system[J]. J Oleo Sci, 2015, 64(4):405-413. [12] 刘美, 程勐, 闫碧涵, 等. 星点设计-效应面法优化丹参酮ⅡA冻干乳的处方[J]. 华西药学杂志, 2015, 30(5):558-560. [13] 梁珍, 张振, 李景果, 等. 伪三元相图联合星点设计效应面法优化益康唑固体脂质纳米粒的处方[J]. 中国药学杂志, 2020, 55(16):1358-1362. [14] 罗国平, 妙苗, 白杨, 等. 伪三元相图法优化复方丁香油微乳制备工艺[J]. 中国药业, 2020, 29(7):87-89. doi: 10.3969/j.issn.1006-4931.2020.07.024 [15] 曾春姣, 陈玲珑, 李跃辉. 正交试验优选猫爪草纳米乳喷雾剂的提取工艺[J]. 湖南中医杂志, 2020, 36(4):150-152. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 6477
- HTML全文浏览量: 1546
- PDF下载量: 57
- 被引次数: 0
