

 下载:
下载:

 下载:
下载:

-
蛋白降解靶向嵌合体(proteolysis targeting chimera, PROTAC)是近年来最受期待的蛋白降解技术[1]。PROTAC是一种异双功能分子,一端与靶蛋白结合,另一端结合E3连接酶形成靶蛋白-PROTAC-E3三元复合物,E3连接酶诱导靶蛋白泛素化,随后被蛋白酶体识别并降解。PROTAC相较传统药物具有诸多优势,如使难成药靶点实现可成药性、大幅增加可用靶点数量、克服耐药性、提高选择性和活性、降低毒副作用等。但是,PROTAC依赖E3连接酶和蛋白酶体,也存在一些固有缺陷,如主要针对细胞质中可溶性蛋白进行降解,不能降解蛋白聚集物和大的蛋白质,对非蛋白物质的降解无能为力[2]。
自噬-溶酶体途径(ALP)是广泛存在于真核细胞中的蛋白降解系统,它是指在自噬关键蛋白LC3参与下,细胞内膜结构形成自噬小体并将底物包裹,随后转运至溶酶体实现底物降解的过程[3]。该机制涉及的底物范围十分广泛,包括蛋白质聚集体、衰老或受损的细胞器、入侵的病原微生物等。ALP与泛素-蛋白酶体途径互为补充,在细胞中发挥重要的生理功能[4-5]。
2019年,复旦大学鲁伯埙团队基于自噬-溶酶体途径首次提出自噬小体绑定化合物(ATTEC)概念。ATTEC是一种双功能分子,同时结合LC3与靶蛋白,将靶蛋白包裹进自噬小体,并在溶酶体中实现降解。基于这一思想,研究人员将小分子库固定在芯片上,筛选以“分子胶”方式将突变型亨廷顿蛋白(mHTT)和LC3蛋白“黏合”的分子,成功获得4个小分子化合物(10O5、ispinesib、AN1-2, 图1)。这些化合物能有效降解mHTT蛋白,并减弱亨廷顿病相关表型[6]。ATTEC分子通过直接连接自噬蛋白LC3,绕过泛素化过程,是一种利用自噬降解靶标最为直接的策略,对于降解不同类型的靶标具有很大的潜力。但是,ATTEC技术尚处于概念验证阶段,急需拓展靶标应用范围,推动技术的不断成熟。
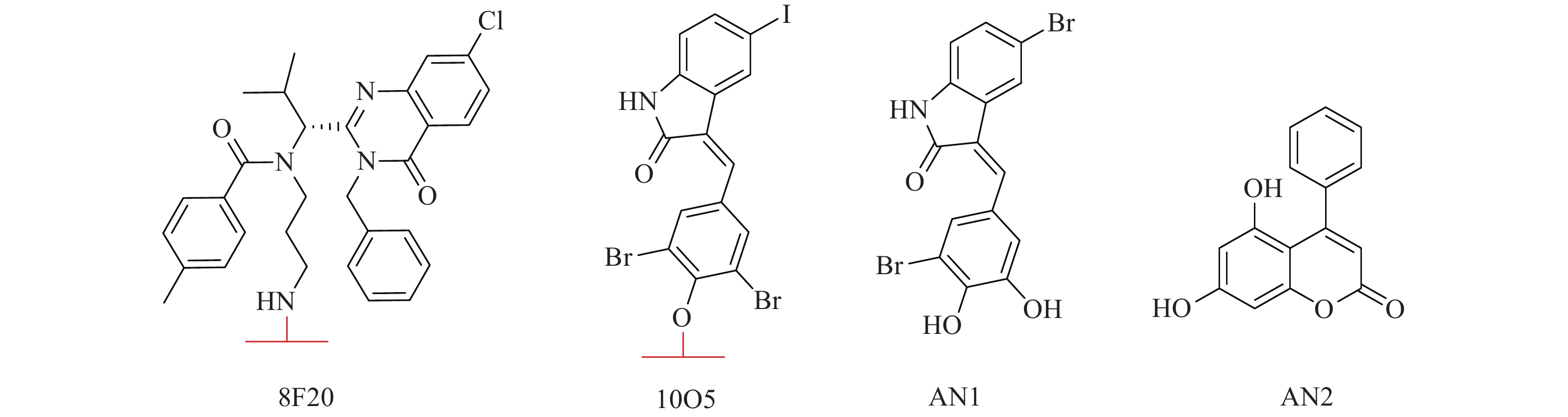
图 1 ATTEC分子胶化学结构
BRD4是BET溴结构域蛋白家族中最重要的成员之一,在人体中广泛分布,对细胞正常生长及细胞周期的调控具有重要意义,与肿瘤发生密切相关,是肿瘤治疗的热门靶点[7],基于PROTAC的BRD4降解剂相继报道[8-9]。BRD4已成为靶向蛋白降解研究的经典体系,为了验证ATTEC降解策略的普适性和可行性,我们以BRD4靶点为研究对象,开展BRD4-ATTEC分子设计、合成和蛋白降解活性评价研究。
-
BRD4-ATTEC分子由三部分组成:BRD4抑制剂、连接子Linker和LC3配体。我们选择经典BRD4抑制剂JQ1作为靶蛋白配体,JQ1与BRD4共晶结构如图2A所示。JQ1的酯基部分暴露于溶剂中,适合作为Linker连接位点,不影响BRD4蛋白结合活性[10];Ispinesib作为LC3配体,由于鲁伯埙团队在进行高通量筛选时,化合物ispinesib的氨基端连接于分子芯片,故选择氨基端作为Linker连接另一位点,不会影响其与LC3蛋白的结合;随后,使用不同长度的烷烃链将两个配体相连设计得到相应目标化合物(图2B、C)。目标化合物通过同时结合BRD4与LC3蛋白,将BRD4靶向至自噬小体中,从而被溶酶体吞噬完成降解。
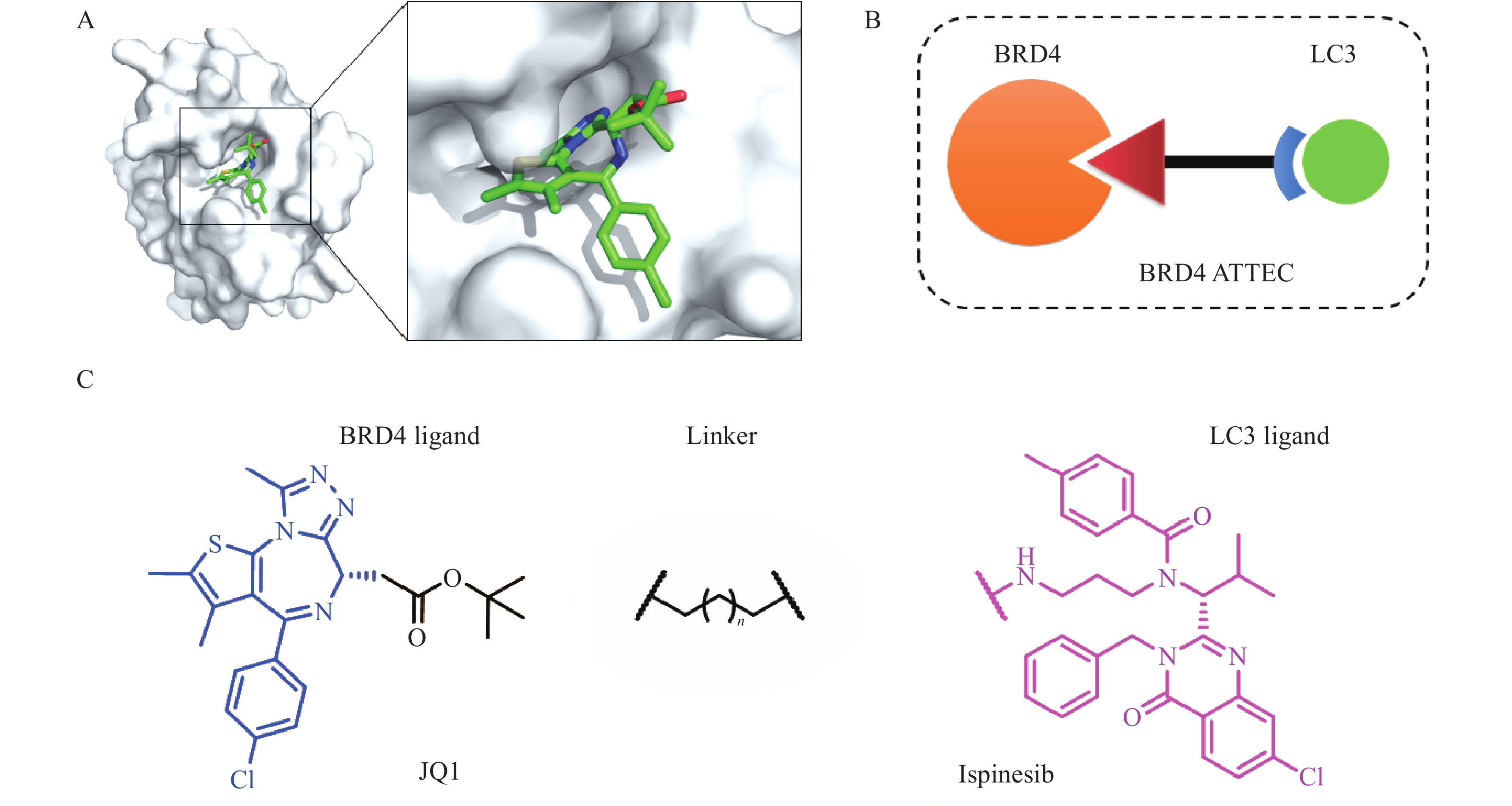
图 2 BRD4 自噬降解设计
-
化学原料均为市售分析纯;免抗BRD4抗体(Abcam,ab128874);免抗GAPDH抗体(Abcam,ab181602);山羊抗免IgG H&L (Alexa Fluor® 680) (Abcam,ab175773);Bruker AVANCE600(Bruker Company, Germany)核磁共振仪,TMS作为内标,化学位移与偶合常数分别用ppm和Hz表示;Agilent 6538 UHD Accurate-Mass Q-TOF LC/MS高分辨质谱(HRMS)仪;上海申光WRR目视熔点仪;Bioteck Synergy2多功能酶标仪;Biorad ChemiDoc成像仪。
-
化合物4的合成路线见图3。
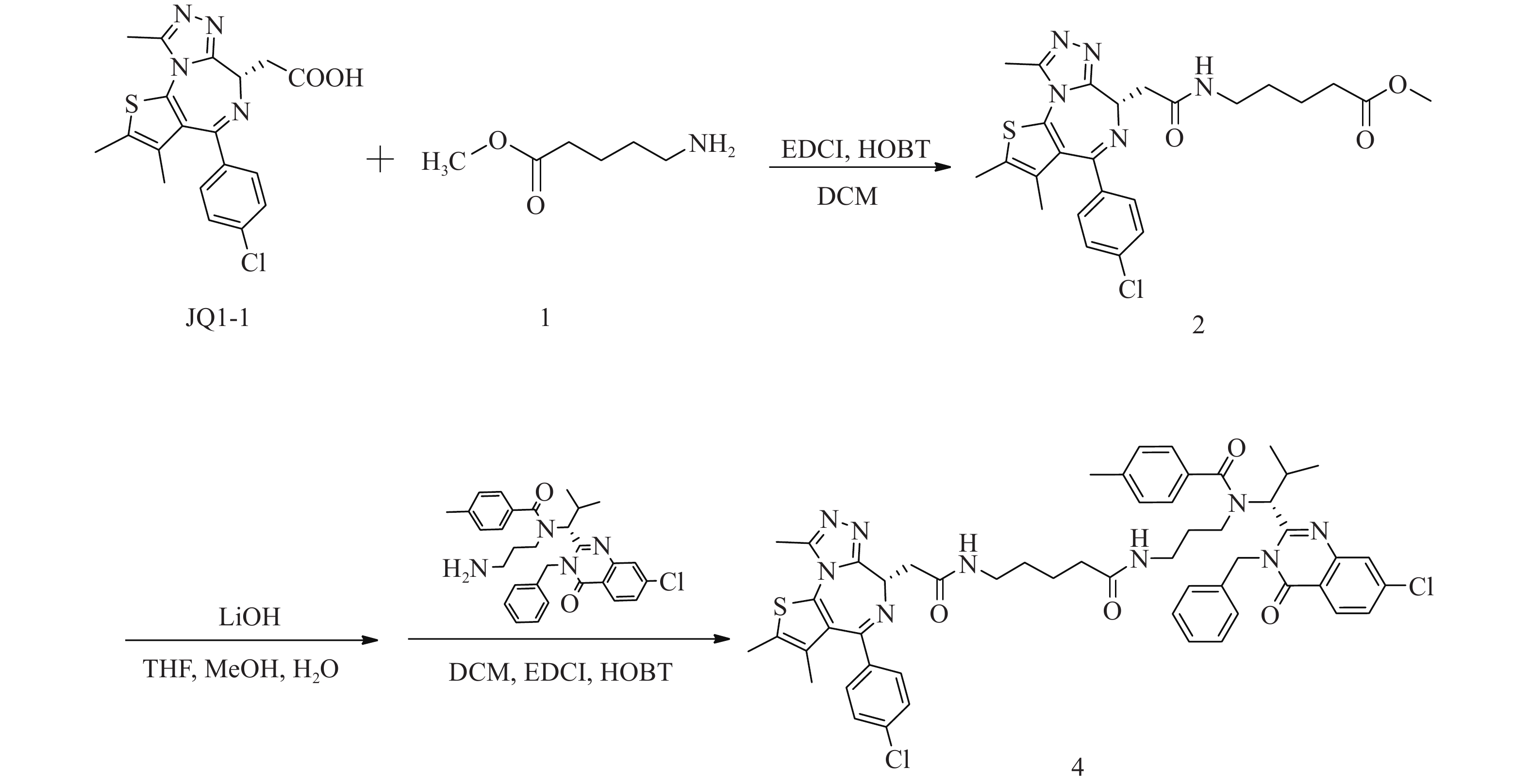
图 3 化合物4合成路线
(S)-5-(2-(4-(4-氯苯基)-2, 3, 9-三甲基-6H-噻吩并[3, 2-f][1, 2, 4]三唑并[4, 3-a][1, 4]二氮杂卓-6-基)乙酰氨基)戊酸甲酯(2)的制备:
将JQ1-1(100 mg,0.25 mmol)溶解于二氯甲烷(DCM, 10 ml)中,加入5-氨基戊酸甲酯(1)(39 mg,0.30 mmol)、EDCI(73 mg,0.38 mmol)和HOBT(51 mg,0.38 mmol),室温下反应8 h。反应完后,加水(200 ml)稀释,并用DCM(50 ml×3)萃取,收集有机层,使用无水硫酸钠干燥,蒸干溶剂,硅胶柱色谱分离(DCM∶MeOH = 98∶2),得淡黄色油状液体(2)91 mg,产率71%;1H NMR (600 MHz, DMSO−d6) δ: 8.20 (t, J=5.7 Hz, 1 H), 7.48 (d, J=8.8 Hz, 2 H), 7.42 (d, J=8.6 Hz, 2 H), 4.50 (dd, J=8.4, 5.7 Hz, 1 H), 3.57 (s, 3 H), 3.28−3.22 (m, 1 H), 3.19−3.12 (m, 2 H), 3.10−3.03 (m, 1 H), 2.59 (s, 3 H), 2.41 (d, J=0.6 Hz, 3 H), 2.33 (t, J=7.4 Hz, 2 H), 1.62 (s, 3 H), 1.59−1.53 (m, 2 H), 1.48−1.42 (m, 2 H)。
N-((R)-1-(3-苄基-7-氯-4-氧代-3, 4-二氢喹唑啉-2-基)-2-甲基丙基)-N-(3-(5-(2-((S)-4-(4-氯苯基)-2, 3, 9-三甲基-6H-噻吩并[3, 2-f][1, 2, 4]三唑并[4, 3-a][1, 4]二氮杂卓-6-基)乙酰胺基)戊酰胺基丙基)-4-甲基苯甲酰胺(4)的制备:
将化合物2(91 mg,0.18 mmol)溶于THF-MeOH-H2O(3∶2∶1)混合溶剂(6 ml),加入LiOH(17 mg,0.72 mmol),室温反应5 h后蒸干溶剂,使用1 mol/L稀盐酸调pH至6,过滤,收集固体并干燥,得白色固体49 mg(0.10 mmol);将所得白色固体(49 mg,0.10 mmol)溶于DCM(15 ml),加入EDCI(29 mg,0.15 mmol)、HOBT(20 mg,0.15 mmol)和ispinesib(0.10 mmol,52 mg),室温反应8 h后,加水(300 ml)稀释,用DCM(100 ml×3)萃取,收集有机层,使用无水硫酸钠干燥,蒸干溶剂,C18反相柱色谱分离(MeOH∶H2O=63∶37),得白色固体(4)59 mg(0.06 mmol),两步收率33%;1H NMR (600 MHz, DMSO−d6) δ: 8.24 (dd, J=8.6, 3.9 Hz, 1 H), 8.17 (t, J=5.6 Hz, 1 H), 7.81 (t, J=1.7 Hz, 1 H), 7.69−7.65 (m, 1 H), 7.50−7.47 (m, 2 H), 7.42 (d, J=8.3 Hz, 2 H), 7.40−7.35 (m, 3 H), 7.34−7.30 (m, 1 H), 7.29−7.20 (m, 6 H), 5.89 (d, J=16.0 Hz, 1 H), 5.55 (d, J=10.6 Hz, 1 H), 5.06 (d, J=16.3 Hz, 1 H), 4.51 (dd, J=8.1, 5.9 Hz, 1 H), 3.30−3.23 (m, 3 H), 3.20−3.15 (m, 1 H), 3.15−3.09 (m, 1 H), 3.05−2.97 (m, 1 H), 2.79−2.71 (m, 1 H), 2.61 (s, 3 H), 2.58−2.54 (m, 1 H), 2.49 (d, J=7.0 Hz, 1 H), 2.42 (s, 3 H), 2.34 (s, 3 H), 1.87−1.76 (m, 2 H), 1.64−1.60 (m, 3 H), 1.40−1.31 (m, 5 H), 0.91 (d, J=6.4 Hz, 3 H), 0.82−0.87 (m, 1 H), 0.49 (d, J=6.2 Hz, 3 H);13C NMR (151 MHz, DMSO−d6) δ: 172.44, 171.88, 169.76, 163.43, 161.57, 155.70, 155.59, 150.23, 147.64, 139.97, 139.11, 137.22, 137.16, 135.64, 134.25, 132.72, 131.14, 130.57, 130.27, 130.02, 129.34, 129.11, 128.91, 128.48, 127.88, 127.11, 126.89, 126.33, 119.55, 59.44, 54.35, 45.61, 42.90, 38.74, 38.11, 36.17, 35.36, 30.71, 29.33, 28.81, 23.06, 21.36, 19.95, 18.61, 14.48, 13.13, 11.75;HRMS(ESI) m/z calcd for C54H56Cl2N9O4S (M-H)− 996.3559, found 996.3542;熔程:143.1~146.3 ℃。
化合物5、6合成路线如图4所示。
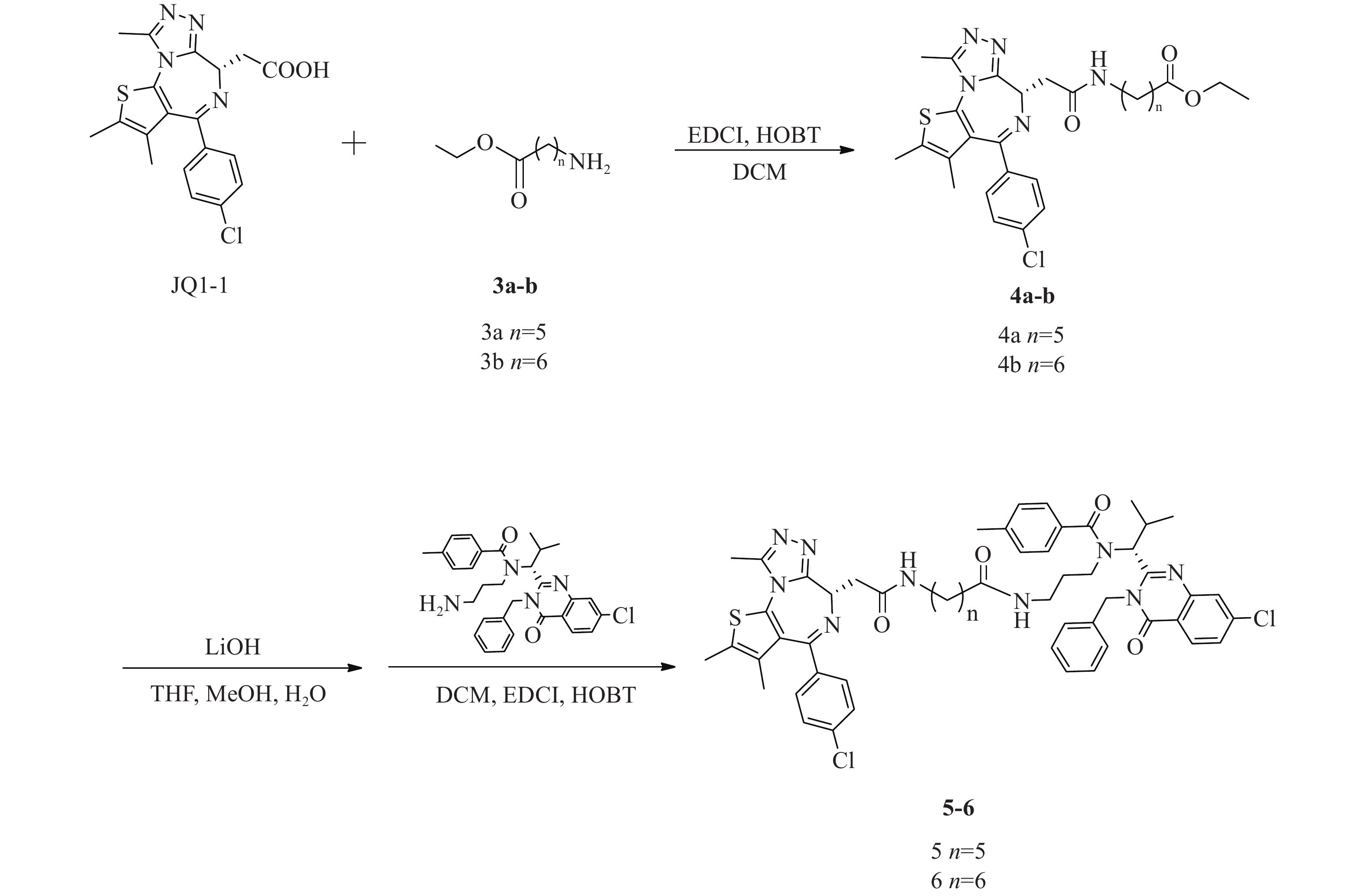
图 4 化合物5和6合成路线
(S)-6-(2-(4-(4-氯苯基)-2, 3, 9-三甲基-6H-噻吩并[3, 2-f][1, 2, 4]三唑并[4, 3-a][1, 4]二氮杂卓-6-基)乙酰氨基)己酸乙酯(4a)的制备:
将JQ1-1(100 mg,0.25 mmol)溶解于DCM(10 ml),加入化合物6-氨基己酸甲酯(3a,44 mg,0.30 mmol)、EDCI(73 mg,0.38 mmol)和HOBT(51 mg,0.38 mmol),室温反应8 h。反应完后,加水(200 ml)稀释,并用DCM(50 ml×3)萃取,收集有机层,使用无水硫酸钠干燥,蒸干溶剂,硅胶柱色谱分离(DCM∶MeOH=98∶2),得淡黄色油状液体(4a)80 mg,产率59%;1H NMR (600 MHz, DMSO - d6) δ∶8.17 (t, J=5.7 Hz, 1 H), 7.48 (d, J=8.8 Hz, 2 H), 7.42 (d, J=8.6 Hz, 2 H), 4.50 (dd, J=8.3, 5.8 Hz, 1 H), 4.04 (q, J=7.2 Hz, 2 H), 3.28−3.22 (m, 1 H), 3.20−3.10 (m, 2 H), 3.08−3.02 (m, 1 H), 2.59 (s, 3 H), 2.41 (s, 3 H), 2.26 (t, J=7.5 Hz, 2 H), 1.62 (s, 3 H), 1.56−1.50 (m, 2 H), 1.47−1.41 (m, 2 H), 1.34−1.27 (m, 2 H), 1.17 (t, J=7.1 Hz, 3 H)。
(S)-7-(2-(4-(4-氯苯基)-2, 3, 9-三甲基-6H-噻吩并[3, 2-f][1, 2, 4]三唑并[4, 3-a][1, 4]二氮杂卓-6-基)乙酰氨基)庚酸乙酯(4b)的制备:
中间体4b的合成步骤参照中间体4a合成,得淡黄色油状液体(4b)99 mg,产率76%;1H NMR (600 MHz, DMSO−d6) δ: 8.16 (t, J=5.7 Hz, 1 H), 7.48 (d, J=8.8 Hz, 2 H), 7.42 (d, J=8.4 Hz, 2 H), 4.50 (dd, J=8.4, 5.9 Hz, 1 H), 4.04 (q, J=7.1 Hz, 2 H), 3.28−3.22 (m, 1 H), 3.20−3.10 (m, 2 H), 3.08−3.02 (m, 1 H), 2.59 (s, 3 H), 2.41 (s, 3 H), 2.26 (t, J=7.4 Hz, 2 H), 1.62 (s, 3 H), 1.54−1.47 (m, 2 H), 1.46−1.39 (m, 2 H), 1.31−1.26 (m, 4 H), 1.16 (t, J=7.1 Hz, 3 H)。
N-((R)-1-(3-苄基-7-氯-4-氧代-3, 4-二氢喹唑啉-2-基)-2-甲基丙基)-N-(3-(6-(2-((S)-4-(4-氯苯基)-2, 3, 9-三甲基-6H-噻吩并[3, 2-f][1, 2, 4]三唑并[4, 3-a][1, 4]二氮杂卓-6-基)乙酰胺基)己酰胺基)丙基)-4-甲基苯甲酰胺(5)的制备:
将化合物4a(80 mg,0.15 mmol)溶于THF-MeOH-H2O(3∶2∶1)混合溶剂(6 ml),加入LiOH(14 mg, 0.6 mmol),室温反应5 h后蒸干溶剂,使用1 mol/L稀盐酸调pH至6,过滤,收集固体并干燥,得白色固体61 mg(0.12 mmol);将所得白色固体(61 mg,0.12 mmol)溶于 DCM(15 ml)中,加入EDCI(35 mg,0.18 mmol)、HOBT(24 mg,0.18 mmol)和化合物ispinesib(0.12 mmol,62 mg),室温下反应8 h后,加水(300 ml)稀释,并用DCM(100 ml × 3)萃取,收集有机层,使用无水硫酸钠干燥,蒸干溶剂,C18反相柱色谱分离(MeOH∶H2O=63∶37),得白色固体(5)(64 mg,0.06 mmol),两步收率42%;1H NMR (600 MHz, DMSO−d6) δ: 8.23 (d, J=8.6 Hz, 1 H), 8.17 (t, J=5.6 Hz, 1 H), 7.79 (d, J=1.8 Hz, 1 H), 7.68−7.65 (m, 1 H), 7.49−7.45 (m, 2 H), 7.42 (d, J=7.9 Hz, 2 H), 7.39−7.34 (m, 3 H), 7.33−7.28 (m, 1 H), 7.28−7.19 (m, 6 H), 5.88 (d, J=16.1 Hz, 1 H), 5.54 (d, J=10.3 Hz, 1 H), 5.05 (d, J=16.5 Hz, 1 H), 4.51 (dd, J=8.2, 6.1 Hz, 1 H), 3.29−3.21 (m, 3 H), 3.21−3.15 (m, 1 H), 3.14−3.07 (m, 1 H), 3.07−3.00 (m, 1 H), 2.77−2.69 (m, 1 H), 2.60 (d, J=1.3 Hz, 3 H), 2.55−2.52 (m, 1 H), 2.50−2.47 (m, 1 H), 2.41 (s, 3 H), 2.33 (s, 3 H), 1.83−1.71 (m, 2 H), 1.62 (s, 3 H), 1.43−1.37 (m, 2 H), 1.36−1.27 (m, 3 H), 1.22−1.15 (m, 2 H), 0.90 (d, J=6.6 Hz, 3 H), 0.87−0.80 (m, 1 H), 0.48 (d, J=5.9 Hz, 3 H); 13C NMR (151 MHz, DMSO−d6) δ: 172.45, 171.96, 169.74, 163.43, 161.57, 155.70, 155.60, 150.24, 147.64, 139.96, 139.11, 137.22, 137.16, 135.67, 134.25, 132.71, 131.16, 130.55, 130.27, 130.03, 129.34, 129.11, 128.90, 128.48, 127.88, 127.13, 126.88, 126.33, 119.55, 59.44, 54.36, 45.62, 42.90, 38.89, 38.08, 36.18, 35.70, 30.69, 29.48, 28.81, 26.61, 25.35, 21.36, 19.95, 18.61, 14.49, 13.13, 11.74;HRMS(ESI) m/z calcd for C55H59Cl3N9O4S (M+Cl)− 1046.3482, found 1046.3443;熔程:143.0~145.2 ℃。
N-((R)-1-(3-苄基-7-氯-4-氧代-3, 4-二氢喹唑啉-2-基)-2-甲基丙基)-N-(3-(7-(2-((S)-4-(4-氯苯基)-2, 3, 9-三甲基-6H-噻吩并[3, 2-f][1, 2, 4]三唑并[4, 3-a][1, 4]二氮杂卓-6-基)乙酰胺基)庚酰胺基)丙基)-4-甲基苯甲酰胺(6)的制备:
化合物6的合成步骤参照化合物5合成,得白色固体(6)(71 mg,0.07 mmol),两步收率36%;1H NMR (600 MHz, DMSO−d6) δ: 8.23 (d, J=8.6 Hz, 1 H), 8.17 (t, J=5.6 Hz, 1 H), 7.79 (d, J=2.0 Hz, 1 H), 7.66 (dd, J=8.6, 2.0 Hz, 1 H), 7.50−7.46 (m, 2 H), 7.44−7.40 (m, 2 H), 7.40−7.34 (m, 3 H), 7.33−7.28 (m, 1 H), 7.27−7.19 (m, 6 H), 5.88 (d, J=16.1 Hz, 1 H), 5.54 (d, J=10.5 Hz, 1 H), 5.05 (d, J=16.3 Hz, 1 H), 4.51 (dd, J=8.3, 6.1 Hz, 1 H), 3.29−3.21 (m, 3 H), 3.21−3.17 (m, 1 H), 3.16−3.09 (m, 1 H), 3.08−3.01 (m, 1 H), 2.77−2.69 (m, 1 H), 2.59 (s, 3 H), 2.57−2.53 (m, 1 H), 2.50−2.46 (m, 1 H), 2.41 (s, 3 H), 2.33 (s, 3 H), 1.84−1.70 (m, 2 H), 1.61 (s, 3 H), 1.44−1.38 (m, 2 H), 1.36−1.28 (m, 3 H), 1.27−1.25 (m, 1 H), 1.23−1.21 (m, 1 H), 1.19−1.12 (m, 2 H), 0.89 (d, J=6.6 Hz, 3 H), 0.87−0.81 (m, 1 H), 0.47 (d, J=6.2 Hz, 3 H);13C NMR (151 MHz, DMSO−d6) δ: 172.45, 172.00, 169.74, 163.44, 161.56, 155.70, 155.60, 150.24, 147.64, 139.95, 139.09, 137.21, 137.16, 135.69, 134.25, 132.71, 131.15, 130.54, 130.26, 130.04, 129.33, 129.11, 128.90, 128.47, 127.88, 127.12, 126.87, 126.32, 119.55, 59.44, 54.37, 45.62, 42.90, 38.90, 38.11, 36.16, 35.67, 30.71, 29.61, 28.89, 28.81, 26.61, 25.53, 21.36, 19.95, 18.60, 14.48, 13.13, 11.74;HRMS(ESI) m/z calcd for C56H61Cl2N9O4SNa (M+Na)+ 1048.3836, found 1048.3892;熔程:142.0~144.7 ℃。
化合物7、8合成路线如图5所示。
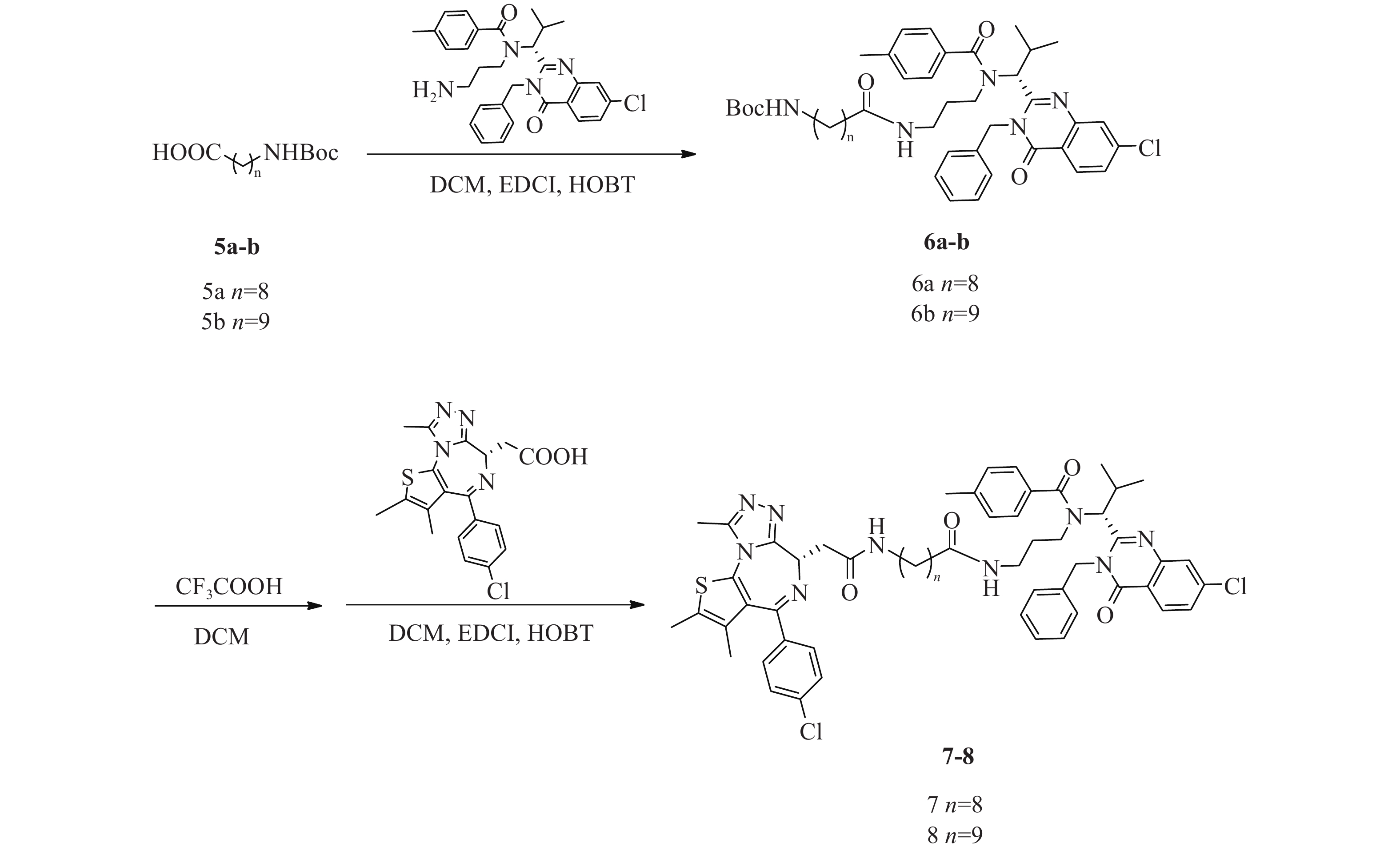
图 5 化合物7、8合成路线
(R)-(9-((3-(N-(1-(3-苄基-7-氯-4-氧代 -3, 4-二氢喹唑啉 -2-基)-2-甲基丙基)-4-甲基苯甲酰胺基)丙基)氨基)-9-氧代壬基)氨基甲酸叔丁酯(6a)的制备:
将ispinesib(100 mg,0.2 mmol)溶解于DCM(10 ml)中,加入化合物9-((叔丁氧基羰基)氨基)壬酸(5a)(66 mg,0.24 mmol),EDCI(58 mg,0.30 mmol)和HOBT(41 mg,0.30 mmol),室温下反应8 h。反应完后,加水(200 ml)稀释,并用DCM(50 ml×3)萃取,收集有机层,使用无水硫酸钠干燥,蒸干溶剂,硅胶柱色谱分离(DCM∶MeOH=98∶2),得淡黄色油状液体(6a)93 mg,产率60%;1H NMR (600 MHz, DMSO−d6) δ: 8.22 (d, J=8.4 Hz, 1 H), 7.77 (d, J=2.0 Hz, 1 H), 7.65 (dd, J=8.5, 2.1 Hz, 1 H), 7.38−7.31 (m, 3 H), 7.31−7.27 (m, 1 H), 7.26−7.19 (m, 6 H), 6.74 (t, J=5.5 Hz, 1 H), 5.88 (d, J=16.1 Hz, 1 H), 5.53 (d, J=10.5 Hz, 1 H), 5.05 (d, J=16.3 Hz, 1 H), 3.26−3.20 (m, 2 H), 2.87 (q, J=6.7 Hz, 2 H), 2.76−2.68 (m, 1 H), 2.55−2.51 (m, 1 H), 2.49−2.45 (m, 1 H), 2.33 (s, 3 H), 1.81−1.69 (m, 2 H), 1.36 (s, 9 H), 1.33−1.27 (m, 4 H), 1.23−1.15 (m, 7 H), 1.14−1.08 (m, 2 H), 0.89 (d, J=6.8 Hz, 3 H), 0.86−0.80 (m, 1 H), 0.47 (d, J=6.2 Hz, 3 H)。
(R)-(10-((3-(N-(1-(3-苄基-7-氯-4-氧代 -3, 4-二氢喹唑啉 -2-基)-2-甲基丙基)-4-甲基苯甲酰胺基)丙基)氨基)-9-氧代壬基)氨基甲酸叔丁酯(6b)的制备:
中间体6b的合成步骤参照中间体6a合成,得淡黄色油状液体(6b)82 mg,产率52%;1H NMR (600 MHz, DMSO−d6) δ: 8.22 (d, J=8.6 Hz, 1 H), 7.78 (d, J=2.0 Hz, 1 H), 7.65 (dd, J=8.6, 2.0 Hz, 1 H), 7.38−7.28 (m, 4 H), 7.26−7.18 (m, 6 H), 6.75 (t, J=5.5 Hz, 1 H), 5.87 (d, J=16.7 Hz, 1 H), 5.53 (d, J=10.5 Hz, 1 H), 5.04 (d, J=16.7 Hz, 1 H), 3.25−3.19 (m, 2 H), 2.87 (q, J=6.4 Hz, 2 H), 2.75−2.68 (m, 1 H), 2.54−2.51 (m, 1 H), 2.49−2.46 (m, 1 H), 2.33 (s, 3 H), 1.81−1.68 (m, 2 H), 1.36 (s, 9 H), 1.34−1.09 (m, 15 H), 0.89 (d, J=6.8 Hz, 3 H), 0.86−0.80 (m, 1 H), 0.47 (d, J=6.2 Hz, 3 H)。
N-((R)-1-(3-苄基-7-氯-4-氧代-3, 4-二氢喹唑啉-2-基)-2-甲基丙基)-N-(3-(9-(2-((S)-4-(4-氯苯基)-2, 3, 9-三甲基-6H-噻吩并[3, 2-f][1, 2, 4]三唑并[4, 3-a][1, 4]二氮杂卓-6-基)乙酰胺基)壬酰胺基)丙基)-4-甲基苯甲酰胺(7)的制备:
将化合物6a(93 mg,0.12 mmol)溶于DCM(3 ml)中,加入CF3COOH(1 ml),室温反应4 h后蒸干溶剂,得无色油状液体73 mg(0.11 mmol);将所得无色油状液体(73 mg,0.11 mmol)溶于DCM(15 ml)中,加入EDCI(31 mg,0.16 mmol)、HOBT(22 mg,0.16 mmol)和化合物JQ1-1(0.11 mmol,44 mg),室温下反应8 h,加水(300 ml)稀释,并用DCM(100 ml×3)萃取,收集有机层,使用无水硫酸钠干燥,蒸干溶剂,C18反相柱色谱分离(MeOH∶H2O=68∶32),得白色固体(7)(53 mg,0.05 mmol),两步收率46%;1H NMR (600 MHz, DMSO−d6) δ: 8.24 (d, J=8.6 Hz, 1 H), 8.19 (t, J=5.5 Hz, 1 H), 7.80 (d, J=1.8 Hz, 1 H), 7.67 (dd, J=8.6, 2.0 Hz, 1 H), 7.50−7.47 (m, 2 H), 7.46−7.42 (m, 2 H), 7.40−7.34 (m, 3 H), 7.34−7.29 (m, 1 H), 7.29−7.21 (m, 6 H), 5.89 (d, J=16.0 Hz, 1 H), 5.55 (d, J=10.6 Hz, 1 H), 5.06 (d, J=16.3 Hz, 1 H), 4.52 (dd, J=8.3, 6.1 Hz, 1 H), 3.30−3.22 (m, 3 H), 3.22−3.17 (m, 1 H), 3.16−3.05 (m, 2 H), 2.77−2.70 (m, 1 H), 2.61 (s, 3 H), 2.58−2.53 (m, 1 H), 2.51−2.47 (m, 1 H), 2.42 (s, 3 H), 2.34 (s, 3 H), 1.84−1.71 (m, 2 H), 1.63 (s, 3 H), 1.48−1.41 (m, 2 H), 1.35−1.25 (m, 7 H), 1.23−1.17 (m, 2 H), 1.17−1.11 (m, 2 H), 0.91 (d, J=6.8 Hz, 3 H), 0.88−0.82 (m, 1 H), 0.49 (d, J=6.2 Hz, 3 H);13C NMR (151 MHz, DMSO−d6) δ: 172.45, 172.02, 169.76, 163.41, 161.56, 155.70, 155.59, 150.23, 147.64, 139.94, 139.10, 137.20, 137.15, 135.68, 134.25, 132.71, 131.16, 130.56, 130.25, 130.03, 129.33, 129.11, 128.87, 128.46, 127.87, 127.12, 126.87, 126.33, 119.55, 59.44, 54.38, 45.63, 42.91, 38.91, 38.13, 36.17, 35.73, 30.69, 29.71, 29.23, 29.14, 28.81, 26.85, 25.57, 21.36, 19.96, 18.61, 14.48, 13.12, 11.74;HRMS(ESI) m/z calcd for C58H66Cl2N9O4S (M+H)+ 1054.433, found 1054.4367;熔程:134.7~139.1 ℃。
N-((R)-1-(3-苄基-7-氯-4-氧代-3, 4-二氢喹唑啉-2-基)-2-甲基丙基)-N-(3-(10-(2-((S)-4-(4-氯苯基)-2, 3, 9-三甲基-6H-噻吩并[3, 2-f][1, 2, 4]三唑并[4, 3-a][1, 4]二氮杂卓-6-基)乙酰胺基)癸酰胺基)丙基)-4-甲基苯甲酰胺(8)的制备:
化合物8的合成步骤参照化合物7合成,得白色固体47 mg,两步收率49%;1H NMR (600 MHz, DMSO−d6)δ: 8.24 (d, J=8.6 Hz, 1 H), 8.18 (t, J=5.6 Hz, 1 H), 7.80 (d, J=1.8 Hz, 1 H), 7.67 (dd, J=8.6, 2.0 Hz, 1 H), 7.50−7.47 (m, 2 H), 7.46−7.42 (m, 2 H), 7.40−7.34 (m, 3 H), 7.34−7.30 (m, 1 H), 7.29−7.21 (m, 6 H), 5.89 (d, J=16.0 Hz, 1 H), 5.55 (d, J=10.6 Hz, 1 H), 5.06 (d, J=16.5 Hz, 1 H), 4.52 (dd, J=8.3, 5.9 Hz, 1 H), 3.30−3.22 (m, 3 H), 3.21−3.16 (m, 1 H), 3.16−3.11 (m, 1 H), 3.11−3.04 (m, 1 H), 2.77−2.70 (m, 1 H), 2.61 (s, 3 H), 2.58−2.53 (m, 1 H), 2.51−2.47 (m, 1 H), 2.42 (s, 3 H), 2.34 (s, 3 H), 1.83−1.71 (m, 2 H), 1.63 (s, 3 H), 1.48−1.41 (m, 2 H), 1.35−1.25 (m, 8 H), 1.22−1.19 (m, 3 H), 1.16−1.09 (m, 2 H), 0.91 (d, J=6.8 Hz, 3 H), 0.89−0.82 (m, 1 H), 0.49 (d, J=6.2 Hz, 3 H);13C NMR (151 MHz, DMSO−d6) δ: 172.45, 172.02, 169.76, 163.40, 161.56, 155.71, 155.60, 150.23, 147.64, 139.93, 139.10, 137.19, 137.15, 135.69, 134.26, 132.72, 131.16, 130.56, 130.25, 130.04, 129.33, 129.11, 128.87, 128.45, 127.87, 127.12, 126.87, 126.34, 119.55, 59.45, 54.39, 45.63, 42.91, 38.93, 38.15, 36.17, 35.74, 30.69, 29.73, 29.40, 29.27, 29.22, 29.15, 28.81, 26.87, 25.57, 21.36, 19.95, 18.62, 14.48, 13.12, 11.74;HRMS(ESI) m/z calcd for C59H68Cl2N9O4S (M+H)+ 1068.4487, found 1068.4488;熔程:132.9~138.5 ℃。
-
将细胞以4×105个/孔的密度接种于6孔板中,培养24 h;根据实验需要,选取相应浓度的PAGE凝胶快速制备试剂盒,取40 μg总蛋白及5 μl蛋白marker上样,恒压120 V电泳90 min;然后恒流300 mA 转膜180 min转至PVDF膜上;转膜结束后,根据marker剪下目的条带,配制无蛋白快速封闭液(5×)封闭30 min;用TBST清洗残留封闭液,用5% BSA稀释相应一抗,4 ℃摇床孵育过夜;用TBST 洗膜3次,每次10min;使用荧光兔二抗,室温孵育1 h; 用TBST 洗膜3次,每次5 min;最后在Biorad ChemiDoc成像仪下拍照。
-
蛋白印迹法结果表明,Linker长度与化合物降解活性密切相关,Linker长度为4个碳原子长度时(化合物4),BRD4降解活性最强。化合物4在HCT116、MDA-MB-231、Hela和A549四种肿瘤细胞中均表现出下调BRD4蛋白水平的活性,而其他化合物在10 μmol/L与1 μmol/L浓度下对胞内BRD4蛋白水平均无明显影响(图6)。
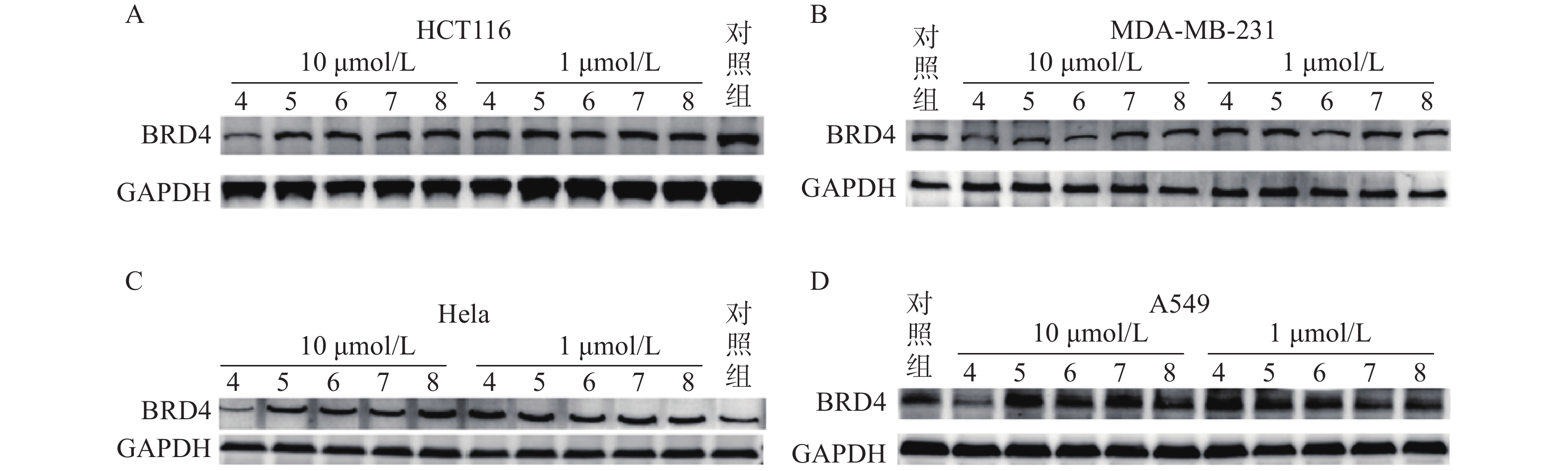
图 6 目标化合物降解BRD4蛋白
-
基于ATTEC策略,我们设计并成功合成5个BRD4-ATTEC分子,所有目标化合物均经过了核磁和质谱结构确证,纯度均高于95%。在蛋白降解活性测试中,我们发现化合物4在10 μmol/L浓度下对4种肿瘤细胞均显示出诱导BRD4蛋白降解活性,表现出广谱有效的特征,为后续BRD4自噬降解的设计和结构优化提供了有效的先导化合物。本研究验证了ATTEC策略能有效诱导BRD4蛋白降解,并获得BRD4自噬降解剂先导化合物,拓展了靶向自噬降解的适用范围,后续的结构优化研究尚在进展之中。
Design, synthesis and degradation activity of BRD4-targeting ATTECs
-
摘要:
目的 基于自噬小体绑定化合物(ATTEC)策略,设计并合成靶向BRD4的自噬降解剂,验证其降解活性。 方法 以化合物伊斯平斯(ispinesib)为LC3配体,通过不同长度的linker与化合物JQ1相连,产物结构经1H NMR、13C NMR与 ESI-MS确证,并使用蛋白印迹法(Western Blot)技术测试其在不同细胞系中诱导BRD4的降解活性。 结果 获得5个首次报道的BRD4-ATTEC分子,化合物 4 在不同细胞系中显示出一定的BRD4降解活性。 结论 本研究发现了新型BRD4自噬降解剂,拓展了靶向自噬降解的适用范围。 Abstract:Objective To design and synthesize autophagic degraders targeting BRD4 based on autophagosome tethering compound (ATTEC) strategy and test their BRD4 degradation activity. Methods BRD4-targeting ATTECs were constructed by conjugating ispinesib that used as a LC3 ligand and JQ1 through a variety of alkane linkers. The final compounds were confirmed by 1H NMR, 13C NMR and ESI-MS, and their degradation activity in different cell lines were tested by Western Blot. Results Five BRD4-ATTEC molecules were successfully synthesized for the first time. Compound 4 showed moderate BRD4 degradation activity in different cell lines. Conclusion The novel BRD4 autophagic degraders were discovered, which expanded the applicability of targeted autophagic degradation via ATTEC. -
Key words:
- autophagosome-tethering compounds /
- BRD4 /
- LC3 /
- Ispinesib
-
河豚毒素(TTX)是一种强效生物毒素,是目前已知毒性最强的生物毒素之一,主要存在于河豚鱼和部分海洋生物中。据报道,河豚毒素能与电压门控钠离子通道1∶1结合,阻断钠离子内流,从而发挥抑制兴奋的作用[1]。基于其对钠离子通道不同亚型的阻断,河豚毒素在一定剂量范围可以起到镇痛、局麻等多种药用效果[2-6]。其中镇痛效用为当前主要研究领域,相较于临床常用的阿片类药物,河豚毒素发挥镇痛作用无成瘾性,副作用小,肝肾功能损害小,但其治疗剂量极低,且治疗窗口狭窄,低剂量往往带来频繁的给药次数,要想良好的发挥镇痛作用,就需要选取合适的剂型来更好的发挥其作用。微球作为长缓释制剂的优良载体,经常包载治疗窗狭窄、半衰期较短的药物以发挥长周期药效的作用,为测定微球中TTX含量等数据,需要一套适用的检测方法。
目前针对河豚毒素定量检测的方法多为生物样本检测,应用于动物体内河豚毒素检测和人河豚毒素中毒血液检测,针对河豚毒素的体外测定方法较少,药用制剂的检测方法更是稀少。本方法的建立适用于河豚毒素药用制剂中的含量检测,为河豚毒素药用开发的含量测定提供了新选择。主流的河豚毒素含量测定方法包括小鼠生物法[7-8]、免疫测定法[9-11]、高效液相色谱法(HPLC)[12-13]、液相色谱-质谱联用法[14-16]、气相色谱-质谱联用法[17]等。高效液相色谱法因其适用性广、稳定性好及灵敏度良好而备受欢迎。但由于河豚毒素不溶于任何有机溶剂,仅溶于弱酸水溶液,常见的方法难以较好地保留河豚毒素。本研究使用庚烷磺酸钠作为离子对试剂,通过河豚毒素与离子对试剂结合形成复合分子,提高其在色谱柱上的保留能力,从而更好地分离河豚毒素与其他物质 [18-19]。
根据河豚毒素的作用功效和缓释长效镇痛目标,本研究制备了河豚毒素缓释微球。为考察微球中河豚毒素的含量,需要建立相应的含量测定方法。因此,本研究采用HPLC方法建立针对河豚毒素缓释微球的含量分析方法,以期为河豚毒素微球中的含量测定提供依据。
1. 仪器与试药
1.1 仪器
LC-20AD高效液相色谱仪(日本岛津制作所);电子天平(METTLER TOLEDO,瑞士);数显pH计(sartorius,德国),紫外可见分光光度计[安捷伦科技(中国)有限公司];循环水式真空泵(上海豫康科教仪器设备有限公司);SECURA125-1CN型十万分之一电子天平(赛多利斯,德国);Arium@ mini超纯水机(赛多利斯,德国)。
1.2 药品与试剂
河豚毒素标准品(98%,中洋生物科技股份有限公司);PBS缓冲液(武汉普诺赛生物科技有限公司);聚乳酸羟基乙酸共聚物(PLGA, RG 503H,sigma-Aldrich Company);甲酸(色谱纯,国药集团);三氟乙酸(色谱纯,sigma-Aldrich Company);氢氧化钠(分析纯,国药集团化学试剂有限公司);泊洛沙姆-188(sigma-Aldrich Company);叠氮钠(Sigma-Aldrich Company);纯净水(杭州娃哈哈集团有限公司);乙腈(色谱纯,sigma-Aldrich Company);二氯甲烷(色谱纯,国药集团化学试剂有限公司);河豚毒素-PLGA微球、空白PLGA微球(海军军医大学药剂学教研室提供)。
2. 方法与结果
2.1 色谱条件
色谱柱:Agilent ZORBAX SB C18柱(4.6 mm×150 mm,5 μm);流动相:8 mmol/L庚烷磺酸钠(0.005%TFA,1 mol/L NaOH调节pH4.0)水溶液∶乙腈=95∶5;检测波长:200 nm;流速:1.0 ml/min;柱温:30 ℃;分析时间:12 min;进样量:20 μl。
2.2 溶液的制备
2.2.1 对照品溶液的制备
精密称取叠氮钠、泊洛沙姆-188适量,加入磷酸盐缓冲液配制成含0.02%NaN3、0.02%F-68的释放介质,取10 mg河豚毒素标准品,用6 ml 0.1%甲酸溶液溶解,以PBS介质(0.02%NaN3、0.02%F-68)定容得100 μg/ml的对照品溶液。
2.2.2 供试品溶液的制备
精密称取20.00 mg河豚毒素冻干微球,加入1 ml二氯甲烷(DCM)超声使完全溶解,加入经甲酸调节pH至4.0的释放介质,超声后充分振摇,取上层水相溶液过0.22 μm水系滤膜作为供试品溶液。
2.3 色谱系统适应性考察
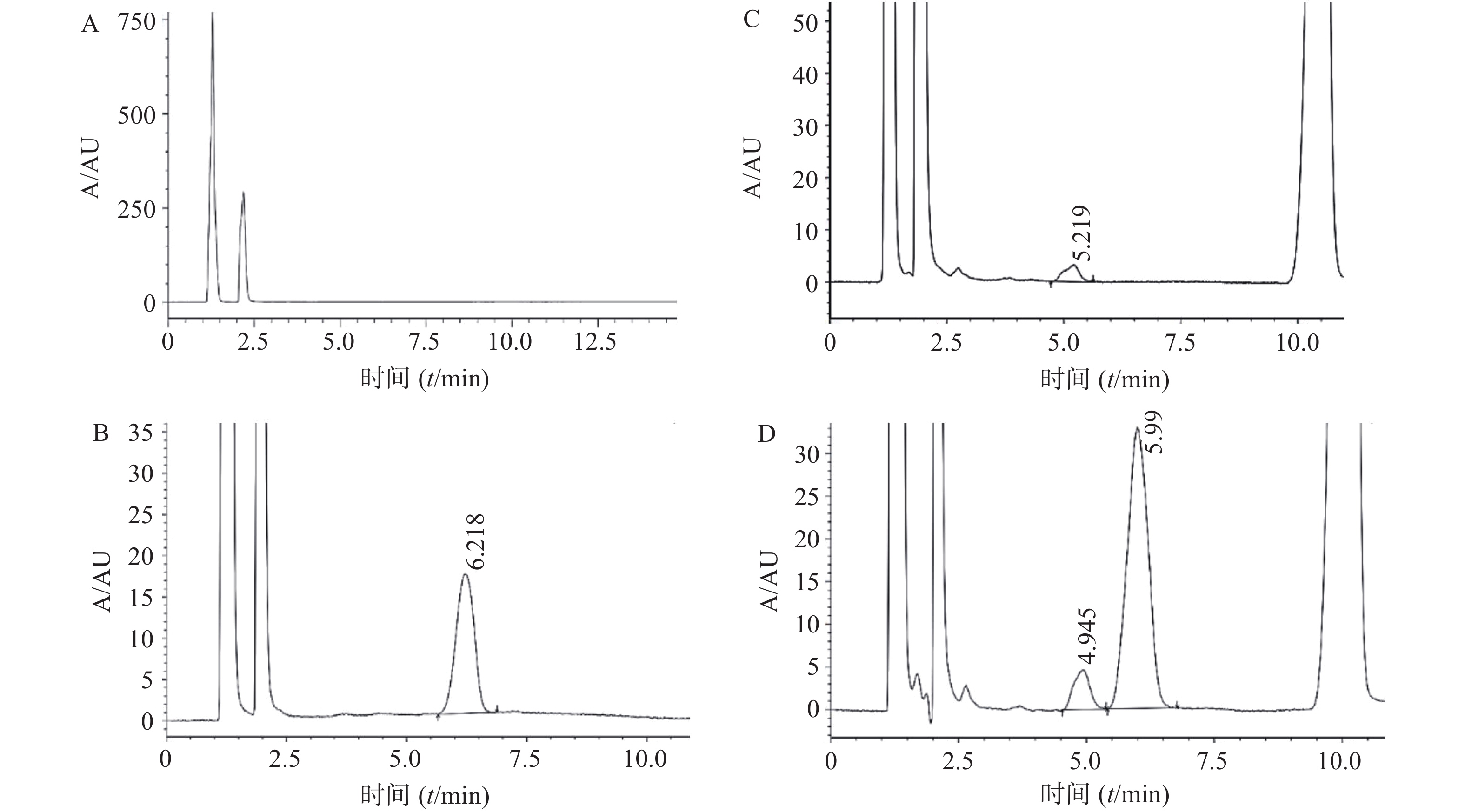
配制以下样品,考察方法专属性:① 1ml PBS释放介质加入50 μl 1%甲酸,为空白释放介质;② 甲酸调节pH的PBS介质稀释得的10 μg/ml TTX标准品溶液;③ 精密称取10 mg空白微球于5 ml EP管,加入1 ml二氯甲烷超声2 min使完全溶解,加入1 ml PBS介质及50 μl 1%甲酸,充分振摇后超声10 min,取上层水相过0.22 μm水系膜,为空白微球样品;④ 精密称取10 mg 空白微球于5 ml EP管,准确加入10 μg/ml TTX标准液1ml,加入1 ml二氯甲烷超声2 min使完全溶解,甲酸调节pH的PBS溶液提取,过膜处理后为河豚毒素微球样品。按照“2.1”项下方法进样检测,图谱如图1。实验结果表明,该方法专属性良好,河豚毒素得到了较好的分离。
2.4 标准曲线的绘制
精密量取对照品溶液于10 ml量瓶,加入PBS介质,分别稀释得20、15、12、10、5、2、1 μg/ml的系列浓度标准液。按照“2.1”项下方法进样检测,记录TTX峰面积。以峰面积(A)为纵坐标,河豚毒素的浓度(Xr μg/ml)为横坐标进行线性回归,得回归方程为Y=22 216X+591.8,r=0.999 9,证明本方法在1~20 μg/ml浓度范围内线性良好。
2.5 精密度试验
由对照品溶液配制低、中、高三个浓度的河豚毒素标准液,分别为2、10、20 μg/ml,进行日内精密度及日间精密度测定。日内精密度测定方法为样品测定5次,计算日内相对偏差;日间精密度测定法为3个浓度样品连续测定5 d,计算日间相对偏差。3个浓度由低到高的日内精密度RSD值分别为0.81%、0.43%、0.58%,日间精密度RSD值分别为1.32%、1.10%、0.68%,均小于2.0%,符合精密度要求。
2.6 重复性试验
按照“2.2.2”项下方法,平行制备5份河豚毒素供试品溶液,按照“2.1”项下色谱条件进行测定。结果显示,RSD值为1.49%,表明该方法重复性良好。
2.7 加样回收率试验
选取低、中、高3个浓度标准溶液,分别为2、10、20 μg/ml,称取10 mg空白微球于5 ml EP管,加入1 ml 二氯甲烷超声溶解和1 ml 标准溶液及50 μl 1%甲酸,充分振摇后超声10 min,超声后取上层水相过膜进样检测,计算回收率,结果见表1。
表 1 河豚毒素加样回收率试验结果(n=3)加入量(μg/ml) 测得量(μg/ml) 回收率(%) RSD(%) 2 1.96 1.98 1.99 98.88±0.82 0.83 10 10.04 9.96 10.00 100.00±0.43 0.43 20 20.04 20.18 20.08 100.49±0.37 0.37 试验结果表明,低、中、高3个浓度的回收率均在98.0%~102.0%之间,3组不同浓度的RSD均小于2%,符合方法学要求。
2.8 微球中药物含量测定
为了考察该方法能否测定微球中还未释放的河豚毒素含量,本实验进行微球中药物含量测定。精密称取河豚毒素-PLGA微球20 mg,加入0.5 ml DCM超声溶解,再加入1%甲酸调节pH至4的PBS溶液2.5 ml,充分振摇后超声10 min,取上清液过膜后进样检测。测定河豚毒素浓度为2.52 μg/ml,换算后计算微球包封率(EE)及载药量(DL),计算方法如下:
$$ \begin{split} &\text{包封率}({\%})=\frac{\text{微球内药物量}}{\text{投入总药物量}}\times 100\\&{\text{载药量}}({\%})=\frac{\text{微球中包含药物量}}{\text{微球总重量}}\times 100 \end{split}$$ 换算可得微球包封率为60.77%,载药量为0.024%。因此,通过此方法可以计算微球的载药量和包封率,为下一步微球释放情况考察时通过测定微球中未释放含量从而间接测定微球的释放量提供依据。
2.9 TTX在释放介质(pH=7.4)中的稳定性
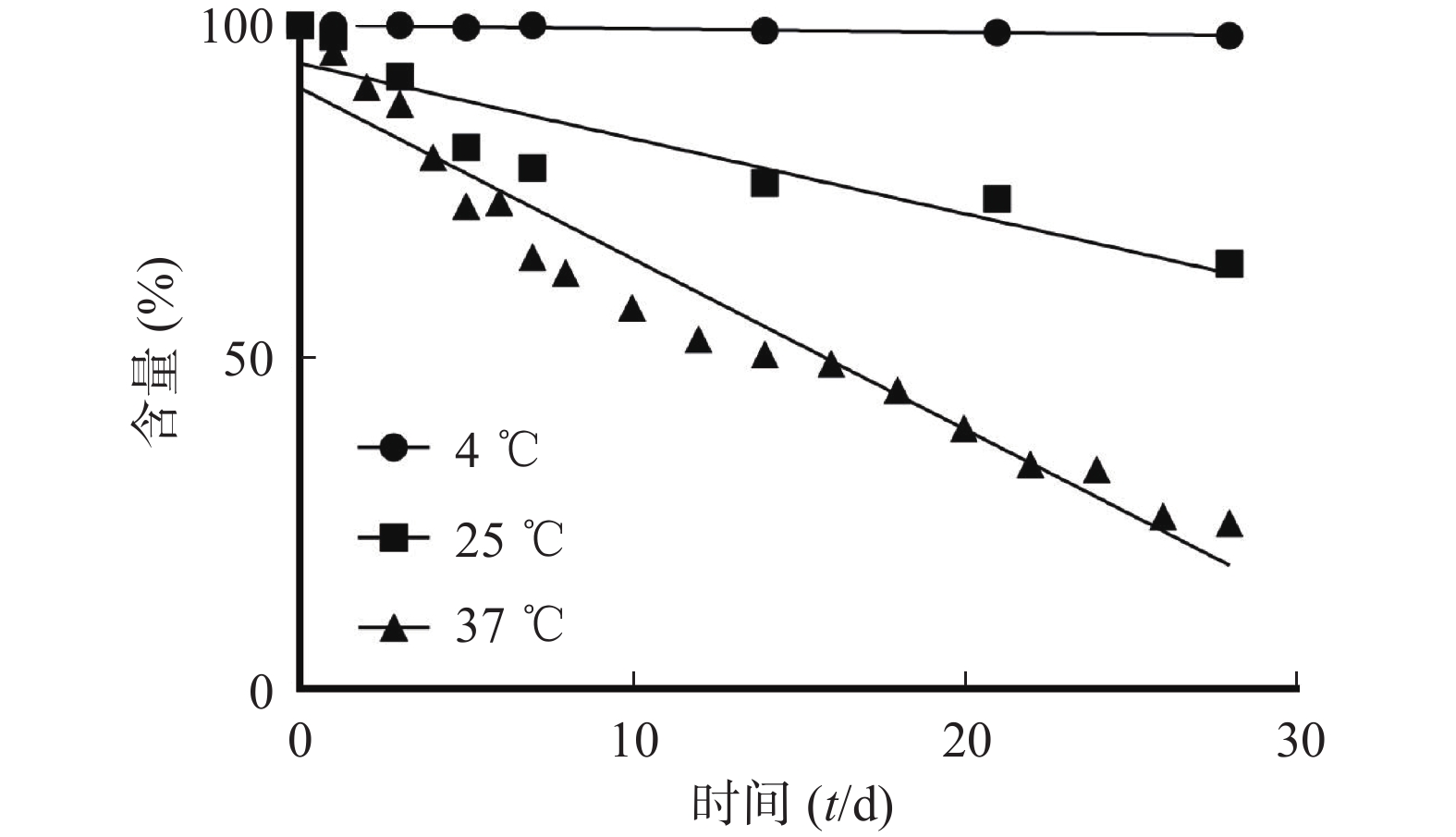
由于河豚毒素溶液受温度影响较大,温度越高,河豚毒素降解越快,在微球长期释放过程中,已释放的河豚毒素发生降解会影响体外释放的测定,故对河豚毒素标准品溶液进行4、25、37 ℃条件下释放介质中的稳定性考察,样品pH均为7.4。4 ℃和25 ℃样品分别在第1、3、5、7、 14、 21、28天取样检测;37 ℃样品在第1周每天取样,其后隔天取样。取12 μg/ml TTX标准品溶液分别置于4、25、37 ℃,100 r/min条件下考察,用于测定TTX在体外释放条件下稳定性,结果见图2。
结果表明,河豚毒素在水溶液中稳定性受温度影响比较大。28 d考察中,河豚毒素在4 ℃放置降解约1.53%,25 ℃放置降解约32.67%,37 ℃放置降解约74.96%。所以,在河豚毒素微球释放测定时不能直接测定释放介质中的河豚毒素含量,而应测定微球中还未释放的河豚毒素含量,从而间接测定微球的释放。
3. 讨论
目前针对河豚毒素定量检测的方法多为生物样本检测,应用于动物体内河豚毒素检测和人河豚毒素中毒血液检测,针对河豚毒素的体外测定方法较少,药用制剂的检测方法更是稀少。本方法的建立适用于河豚毒素药用制剂中的含量检测,为河豚毒素药用开发的含量测定提供了借鉴。
针对河豚毒素微球的含量测定,我们尝试了许多方法,发现常规的反相色谱法对这类物质分离度不高,而反相离子对色谱法分离效果好。为了取得更佳的分离效果,分别考察了Agilent Zorbax SB-C8柱(4.6mm×150mm, 5μm)、shim-pack GIST C18-AQ(4.6mm×250mm, 5μm)、Agilent Zorbax SB C18柱(4.6mm×150mm, 5μm)等不同色谱柱对河豚毒素的分离效能及峰型的影响。结果显示,当色谱柱为Agilent Zorbax SB C18柱时,河豚毒素的分离效果及峰形最佳。
在离子对色谱法中,河豚毒素的分离及峰型等受到多种因素影响,在该方法建立过程中,我们考察了流动相pH、流动相比例等条件对河豚毒素分离效果的影响。流动相pH:我们考察了流动相中pH3.0、4.0、4.5和5.0,不同pH对其峰型有一定的影响,对比筛选后,我们确定了pH4.0时为最佳峰型。流动相比例:流动相比例对TTX出峰时间存在较大的影响,我们考察了90∶10、92∶8、94∶6、95∶5、98∶2等比例,保留时间在4~25 min不等,在保证出峰完整的情况下,调整进样时间至适宜,最终确定比例为95∶5。
微球中的河豚毒素提取我们尝试了不同的有机溶剂破乳,其中包括二氯甲烷、三氯甲烷、乙腈等有机溶剂,最终选用速度最快、溶解最完全的二氯甲烷溶剂破乳提取。
结果证明选用本方法测定缓释微球中的河豚毒素在一定浓度范围内线性良好,专属性强,精密度和回收率均符合方法学要求,可以作为TTX微球含量、释放量的测定方法。
-
图 2 BRD4 自噬降解设计
A. BRD4蛋白与JQ1共晶结构(PDB: 3MXF); B. BRD4-ATTEC自噬降解剂原理; C. BRD4-ATTEC化合物设计
-
[1] SAKAMOTO K M, KIM K B, KUMAGAI A, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation[J]. Proc Natl Acad Sci USA,2001,98(15):8554-8559. doi: 10.1073/pnas.141230798 [2] LU K F, DEN BRAVE F, JENTSCH S. Pathway choice between proteasomal and autophagic degradation[J]. Autophagy,2017,13(10):1799-1800. doi: 10.1080/15548627.2017.1358851 [3] MIZUSHIMA N, LEVINE B, CUERVO A M, et al. Autophagy fights disease through cellular self-digestion[J]. Nature,2008,451(7182):1069-1075. doi: 10.1038/nature06639 [4] TAKAHASHI D, MORIYAMA J, NAKAMURA T, et al. AUTACs: cargo-specific degraders using selective autophagy[J]. Mol Cell,2019,76(5):797-810.e10. doi: 10.1016/j.molcel.2019.09.009 [5] NAKAGAWA I, AMANO A, MIZUSHIMA N, et al. Autophagy defends cells against invading group A Streptococcus[J]. Science,2004,306(5698):1037-1040. doi: 10.1126/science.1103966 [6] LI Z Y, WANG C, WANG Z Y, et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds[J]. Nature,2019,575(7781):203-209. doi: 10.1038/s41586-019-1722-1 [7] GHOSHAL A, YUGANDHAR D, SRIVASTAVA A K. BET inhibitors in cancer therapeutics: a patent review[J]. Expert Opin Ther Pat,2016,26(4):505-522. doi: 10.1517/13543776.2016.1159299 [8] LU J, QIAN Y M, ALTIERI M, et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4[J]. Chem Biol,2015,22(6):755-763. doi: 10.1016/j.chembiol.2015.05.009 [9] ZENGERLE M, CHAN K H, CIULLI A. Selective small molecule induced degradation of the BET bromodomain protein BRD4[J]. ACS Chem Biol,2015,10(8):1770-1777. doi: 10.1021/acschembio.5b00216 [10] FILIPPAKOPOULOS P, QI J, PICAUD S, et al. Selective inhibition of BET bromodomains[J]. Nature,2010,468(7327):1067-1073. doi: 10.1038/nature09504 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3759
- HTML全文浏览量: 1439
- PDF下载量: 42
- 被引次数: 0
