

 下载:
下载:

 下载:
下载:

-
肝纤维化是肝脏对慢性肝损伤的过度修复反应,是一个动态可逆的过程。阻断肝纤维化持续进展是防治肝病向肝硬化甚至肝细胞癌发展的重要策略。目前研究认为,肝星状细胞(HSCs)转化为活化的肌成纤维细胞(MFBs)是细胞外基质 (ECM)的主要来源,也是肝纤维化的主要驱动力[1]。除此之外,大量研究发现,肝巨噬细胞在肝纤维化过程中同样发挥重要作用。
-
肝巨噬细胞是肝脏中一个高度异质的非实质细胞群,占肝脏细胞的10%~15%,主要由枯否细胞 (KCs) 和单核细胞衍生的各种浸润巨噬细胞 (MoMFs) 组成,具有显著异质性。巨噬细胞的异质性以多种来源、多种细胞类型特异性标志物和极化表型为特征。此外,肝巨噬细胞具有显著的可塑性,能够快速响应组织环境的变化,呈现不同的细胞表型。肝巨噬细胞被认为是肝脏抵御病原体的第一道防线,参与包括炎症反应、纤维化形成以及纤维化消退在内的肝纤维化所有阶段[2]。因此,肝内巨噬细胞是肝纤维化的核心调控者,肝纤维化治疗的重要靶细胞。
-
KCs是体内最大的常驻巨噬细胞群,包括卵黄囊来源的KCs和骨髓来源的KCs。KCs可以识别内源性细胞碎片和外源性病原体,感知肝脏损伤进而发生活化。因此,KCs对于肝脏稳态的维持、免疫反应的启动和肝损伤的恢复至关重要。
-
MoMFs充当免疫反应协调者并补充巨噬细胞群以维持肝脏中的稳态。在小鼠体内,MoMFs分为两个亚群:Ly6Chi和Ly6Clo单核细胞。Ly6Chi单核细胞参与炎症反应并产生促炎作用,而Ly6Clo单核细胞则具有组织修复功能。
-
在肝损伤早期,损伤或死亡的肝细胞能够释放损伤相关分子模式 (DAMP),并与肝巨噬细胞相互作用,从而促进肝巨噬细胞的活化、极化和募集。活化的肝巨噬细胞能够分泌大量促炎因子和促纤维化细胞因子,参与炎症反应并促进HSCs的活化和纤维化应答[3](图1)。
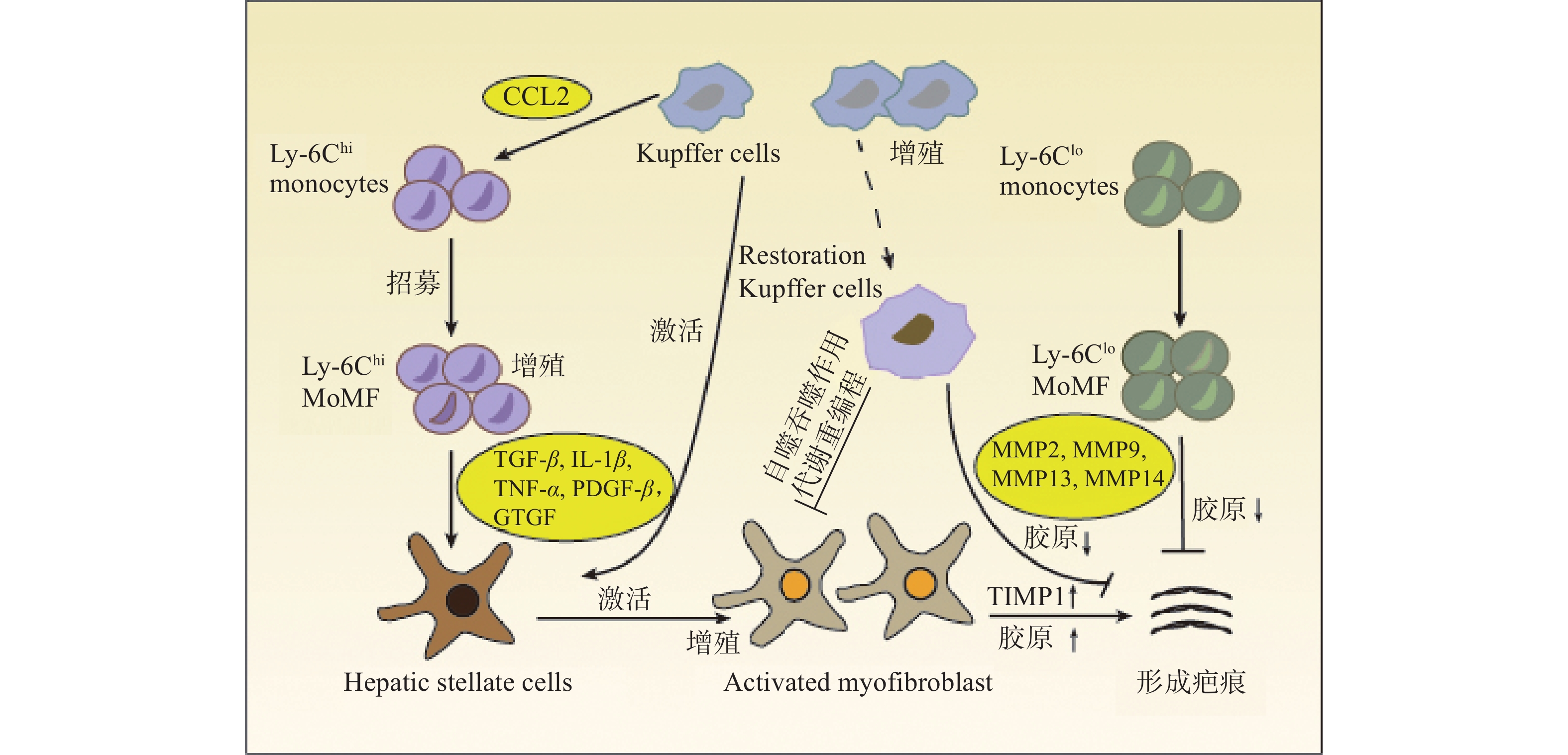
图 1 肝巨噬细胞对肝纤维化的影响
-
HSCs位于肝细胞和肝窦内皮细胞之间的窦周间隙,在正常和受损的肝组织中具有不同的生理功能。正常的生理条件下,HSCs被称为静息的HSCs (qHSCs),负责储存肝脏中的维生素A。当肝损伤后,qHSCs被炎症介质激活,进而分化为 MFBs,分泌大量的ECM并产生纤维瘢痕,导致肝纤维化[4]。肝巨噬细胞能够与HSCs相互作用,重塑免疫微环境和ECM,该相互作用对于肝脏炎症应答和纤维化应答至关重要。
肝巨噬细胞能够分泌与HSCs活化有关的各种细胞因子和趋化因子,如转化生长因子β (TGF-β),肿瘤坏死因子α (TNF-α),白细胞介素1β (IL-1β) 和趋化因子配体2 (CCL2) 等。肝巨噬细胞产生的IL-1β和TNF-α能够激活NF-κB信号通路,增强MFBs的增殖[5]。激活素A (ACTA) 是TGF-β超家族成员之一,能够促进KCs中TNF-α和TGF-β1的表达,诱导HSCs转化为促纤维化表型,提高α-平滑肌肌动蛋白 (α-SMA) 表达水平和迁移能力[6]。此外,肝脏在受到丙型肝炎病毒 (HCV) 感染后,肝巨噬细胞能够分泌趋化因子配体5 (CCL5) ,引起HSCs中炎症小体和纤维化标志物α-SMA与TGF-β的激活[7]。
除细胞因子和趋化因子外,肝巨噬细胞还能够产生其他与HSCs串扰的分子。例如,MoMFs能够通过分泌颗粒蛋白激活qHSCs,促进其转化为MFBs,引起ECM的积累和肝纤维化的形成[8]。HMGB1是一种响应组织损伤而产生的DAMP,主要由肝细胞和KCs分泌,研究表明,HMGB1能够与HSCs中的晚期糖基化终末产物受体(RAGE)结合,激活pMEK1/2、pERK1/2和p-c-Jun通路,增加Ⅰ型胶原蛋白的沉积[9]。
此外,HSCs也可作用于肝巨噬细胞,影响其功能。紫藤凝集素阳性Mac-2结合蛋白 (WFA+-M2BP) 是一种肝纤维化的血清指示剂,可由HSCs分泌。据报道,WFA+-M2BP能够促进KCs表达M2BP,而KCs分泌的M2BP反过来又会进一步加强HSCs的激活。由此说明,在肝纤维化的发展过程中,巨噬细胞和HSCs之间存在正反馈调节[10]。
-
巨噬细胞具有显著的异质性,可以在不同的组织微环境中极化成不同的表型,从而发挥不同作用。通常,巨噬细胞极化的表型可以分为经典活化 (M1) 型和选择活化 (M2) 型。M2型可以进一步分为M2a、M2b、M2c和M2d亚型。
在被脂多糖 (LPS) 和干扰素-γ(IFN-γ) 刺激后,M1型巨噬细胞发生活化,分泌大量促炎细胞因子,如IL-1β、TNF-α、诱导型一氧化氮合酶 (iNOS) 等,从而发挥促炎作用。因此,M1型巨噬细胞在肝损伤起始阶段起重要作用,能够释放促炎因子,加剧炎症反应并促进肌成纤维细胞增殖,最终导致肝纤维化[11]。
M2型巨噬细胞的极化主要由白细胞介素4 (IL-4) 和白细胞介素13 (IL-13)等细胞因子诱导发生,能够分泌抗炎因子,如白细胞介素10 (IL-10) 、TGF-β、精氨酸酶1 (Arg-1) 等,具有抑制炎症、促进组织重塑、预防寄生虫感染以及调节免疫等生物学功能。在肝纤维化的发展过程中,M2型巨噬细胞产生抗炎作用,能够促进伤口的愈合和再生。然而,当肝损伤持续存在时,M2型巨噬细胞释放的TGF-β、血小板衍生生长因子 (PDGF) 、血管内皮生长因子 (VEGF) 等生长因子,会促进MFBs的增殖和活化,加重肝纤维化[12]。
调节巨噬细胞极化可以发挥抗肝纤维化作用。脯氨酸-丝氨酸-苏氨酸-磷酸酶相互作用蛋白2 (PSTPIP2) 能够调节STAT1和NF-κB信号通路,抑制M1型巨噬细胞极化,改善肝脏炎症和肝纤维化[13]。白细胞介素22 (IL-22) 能够调节STAT3、Erk、Akt信号通路的传导,促进巨噬细胞从M1型转变为M2型,从而减缓肝纤维化的进展[14]。
此外,肝巨噬细胞和其他细胞群之间的相互作用对肝巨噬细胞的表型转换也至关重要。自然杀伤 (NK) 细胞在调节巨噬细胞极化方面发挥关键作用,研究发现,DX5+NKp46+ NK细胞能够产生IFN-γ促进M1巨噬细胞极化,在预防非酒精性脂肪性肝炎 (NASH) 进展为肝纤维化方面发挥重要作用[15]。中性粒细胞 (PMN) 能够促进巨噬细胞转变为具有肝脏修复功能的巨噬细胞,有助于肝脏炎症和肝纤维化的自发消退[16]。
-
肝巨噬细胞浸润与慢性炎症和肝纤维化密切相关。在趋化因子和相应受体的介导下,巨噬细胞能够募集到损伤部位,参与炎症和肝纤维化的发展。例如,Ly6Chi MoMFs能够依赖趋化因子受体2 (CCR2) 募集到肝损伤区域,发挥促炎和促纤维化作用。微蛋白 (PSMP) 是一种新型趋化因子,能够促进炎性巨噬细胞的浸润,分泌更多的促炎细胞因子,加剧肝纤维化应答[17]。骨髓细胞上表达的触发受体1 (TREM-1) 可促进KCs的募集和浸润以及促炎细胞因子的产生[18]。
阻碍肝巨噬细胞的浸润与募集有助于肝纤维化的消退。配对免疫球蛋白样2型受体α (PILRα) 是一种抑制性受体,主要在骨髓细胞中表达,在炎症过程中能够抑制肝巨噬细胞与PMN的浸润。研究证实,PILRα能够调节整合素信号传导,阻碍巨噬细胞迁移到受损的肝组织,从而减轻肝脏炎症并缓解肝纤维化[19]。
此外,研究还发现一种新的TREM2+ CD9+瘢痕相关巨噬细胞 (SAMacs) 亚群,其来源于MOMFs,在肝纤维化过程中表现出促纤维化表型[20],可能成为未来肝纤维化治疗的重要靶细胞。
-
在肝纤维化进展中,肝细胞能够分泌一系列DAMP和细胞外囊泡 (EVs) ,可与肝巨噬细胞相互作用,诱导巨噬细胞转变为促炎表型。线粒体DNA (mtDNA) 是一种内源性DAMP,能够激活先天免疫反应[21]。肝细胞衍生的mtDNA能够激活NF-κB信号通路,诱导KCs分泌TNF-α和白细胞介素6 (IL-6) ,引起肝脏炎症和纤维化应答[22]。M1型巨噬细胞分泌的EVs能够激活肝细胞中NLRP3炎性体信号通路,而应激的肝细胞可分泌含有微小RNA-192-50 (miR-192-5p) 和血清CD40L配体 (CD40L) 的EVs,促进M1型巨噬细胞极化[23]。因此,肝细胞和巨噬细胞之间通过释放DAMP和EVs相互作用,促进了肝内炎症反应与肝纤维化进展。
-
细胞代谢重编程是细胞为满足能量需求,通过改变代谢模式促进细胞增殖和生长的机制,包括糖代谢、脂代谢、氨基酸代谢等。
肝巨噬细胞的代谢重编程与巨噬细胞极化紧密联系,影响肝纤维化的进展和消退[24]。c-Rel是NF-κB转录因子家族成员之一,参与巨噬细胞代谢重编程。研究发现,c-Rel能够与6-磷酸果糖激酶-2的启动子结合,诱导巨噬细胞极化和HSCs活化,从而加重炎症反应与肝纤维化[25]。此外,肝脏中铁的代谢失调也与晚期肝纤维化有关。肝内铁的积累能够激活MiT/TFE转录因子,促进M1型巨噬细胞的活化,加重肝纤维化[26]。
膜联蛋白A5是膜联蛋白家族的成员之一,能够与M2型丙酮酸激酶(PKM2)相互作用,将肝巨噬细胞中的糖酵解转换为氧化磷酸化,促进巨噬细胞从M1型转换到M2型,从而改善炎症和肝纤维化[27]。因此,调节肝巨噬细胞的免疫代谢是肝纤维化的潜在治疗策略。
-
自噬是将机体中异常表达的蛋白质和受损的细胞器转移到溶酶体中进行降解,对细胞稳态的维持、细胞存活、分化和生长至关重要。大量研究证实,巨噬细胞自噬对肝脏具有保护作用。例如,KCs的自噬能够抑制细胞活性氧 (ROS) 介导的白细胞介素1α (IL-1α) 和IL-1β的分泌,从而缓解肝脏炎症和纤维化[28]。日本血吸虫卵抗原 (SEA) 诱导的巨噬细胞自噬能够抑制肝脏病理的发展[29]。白细胞介素7 (IL-7) 能够通过激活AMP活化蛋白激酶 (AMPK) 抑制SEA诱导的巨噬细胞自噬,促进炎症细胞对肝脏的浸润,增强MFBs活性,从而加重SEA感染引起的肝纤维化[30]。此外,LC3相关的吞噬作用 (LAP) 是一种非典型的自噬形式,能够将Ly6Chi MoMFs变为Ly6Clo MoMFs。研究表明,LAP可抑制全身炎症,发挥抗肝纤维化作用[31]。
吞噬作用是细胞摄取较大固体颗粒或大分子复合体的过程。肝巨噬细胞能够通过吞噬和清除肝脏中死亡的细胞调节肝脏炎症和纤维化。在肝损伤中,巨噬细胞能够吞噬坏死的肝细胞,诱导Wnt3a的表达并激活Wnt通路,从而促进肝再生。肝脏巨噬细胞的吞噬作用,减弱了受损肝细胞中线粒体衍生的DAMP的释放,从而抑制肝脏瘢痕的形成[32]。因此,调节肝巨噬细胞的自噬和吞噬功能可成为一种新的抗肝纤维化策略,值得进一步研究。
-
除了KCs和MoMFs,其他浸润性巨噬细胞群也与肝纤维化有关。SOCS蛋白是巨噬细胞炎症活性的调节因子,在肝纤维化期间,脾巨噬细胞能够通过上调肝巨噬细胞中的SOCS3信号传导来促进CCL2的分泌,从而促进循环单核细胞的浸润,加剧肝纤维化的发展[33]。
-
肝纤维化的发展与多种细胞和分子机制相关。除肝巨噬细胞外,HSCs、肝细胞、肝窦内皮细胞、胆管细胞和脾细胞也参与了肝纤维化的发展,这些细胞之间的相互作用,能够调控细胞内信号传导,从而影响肝纤维化进展和消退。此外,肝内肝窦的形成和重塑是肝纤维化的关键特征,抑制血管生成也能够减缓肝纤维化的进展。
-
肝巨噬细胞在肝损伤、肝纤维化进展和消退中发挥双重作用。目前研究证实,多种药物能够通过调控肝巨噬细胞的功能发挥抗肝纤维化的作用。因此,基于巨噬细胞在肝纤维化中的作用,人们开发了相关的趋化因子抑制剂、细胞通路拮抗剂,期望为肝纤维化提供新的治疗策略。
-
调节巨噬细胞极化可以治疗肝纤维化。例如,槲皮素能够调控Notch1通路,抑制M1型巨噬细胞极化,缓解炎症反应,从而抑制肝纤维化进展[34];壳寡糖能够通过调控JAK2/STAT1和JAK1/STAT6信号通路,抑制巨噬细胞极化为M1型,增加M2型巨噬细胞数量,从而发挥抗肝纤维化作用[35]。此外,研究还发现,在肝纤维化期间,脾切除能够激活ERK1/2信号通路,促进MOMFs转换为抗炎的Ly6CloMOMFs,从而减轻肝脏炎症和肝纤维化应答[36]。
-
抑制巨噬细胞募集和浸润有助于肝纤维化的消退。姜黄素能够通过抑制KCs的激活减少趋化因子分泌,降低Ly6ChiMOMFs的浸润,从而缓解肝纤维化[37]。鉴于CCL2/CCR2和CCL5/CCR5信号通路在巨噬细胞募集中的关键作用,人们研发出了相关的趋化因子受体拮抗剂,如CCR2拮抗剂RS102895、CCR2/CCR5双拮抗剂 (CVC) 。研究发现,在酒精性肝纤维化模型中,CVC能够明显抑制体内巨噬细胞的募集,展现出较好的的抗纤维化活性[38]。
-
巨噬细胞自噬是一种针对慢性肝损伤和纤维化的保护机制,通过诱导巨噬细胞自噬能够治疗肝纤维化。MJN110是一种单酰基甘油脂肪酶 (MAGL) 抑制剂,在CCl4和BDL诱导的肝纤维化模型中,MJN110的干预能够促使巨噬细胞自噬通量和自噬体生物合成增加、减少肝巨噬细胞数量,从而减缓肝纤维化进展,促进肝纤维化消退[39]。
-
大多数基于肝巨噬细胞的疗法仅在肝纤维化的动物模型中进行了评估,而相关的临床研究数据较少。CVC是CCR2和CCR5双重拮抗剂,两项临床实验数据显示,CVC在伴有纤维化的NASH中具有显著的抗纤维化作用,并且耐受性良好[40]。此外,有研究人员在人体上进行了自体巨噬细胞治疗的安全性评估实验,结果表明,该疗法在肝硬化患者中是安全可行的,这为未来研究其在肝硬化和其他纤维化疾病中的疗效提供了依据[41]。
-
肝纤维化是由各种病因所致慢性肝损伤的修复反应,其特征是ECM在肝内的过度沉积。鉴于肝巨噬细胞在调节肝纤维化反应中的关键作用,人们开发出了针对肝巨噬细胞治疗肝纤维化的新策略。基于抑制KCs活化的靶向疗法已被研究,这些疗法主要通过抑制细胞内炎症信号通路,如NF-κB、ASK1、JNK和p38等信号通路,从而治疗肝纤维化[42]。Loomba等人开发了Selonsertib,一种ASK1信号通路的抑制剂,研究证实,Selonsertib对肝细胞代谢和巨噬细胞活化有影响。在一项随机2期试验中,Selonsertib能够降低NASH和肝纤维化患者的肝脏中胶原蛋白含量和小叶炎症程度,并且能够改善肝细胞凋亡和坏死[43]。此外,肝纤维化治疗的重点是减少MoMFs向肝脏的募集。MoMFs向受损肝脏的募集依赖于活化的肝细胞分泌的几种趋化因子,如趋化因子配体1 (CCL1) ,CCL2,CCL5[44]。因此,调节趋化因子的信号传导也是一种治疗策略,这些疗法主要包括针对趋化因子或受体的单克隆抗体、阻止趋化因子结合的受体拮抗剂、适体分子和阻断趋化因子诱导的细胞内信号传导的小分子抑制剂等[5]。研究发现,使用CCR2敲除能够减弱小鼠MoMFs募集,抑制MFBs活化并减轻肝纤维化[45]。此外,MoMFs可分为导致肝脏损伤的Ly6Chi MOMFs和具有肝脏修复功能的Ly6Clo MOMFs。因此,另一种潜在的策略是通过将Ly6Chi MOMFs转换为Ly6Clo MOMFs来恢复正常的肝功能。研究证实,在CCl4诱导的肝纤维化模型和MCD饮食诱导的NASH模型中,CCL2抑制剂mNOX-E36能够抑制Ly6Chi MOMFs的早期流入,同时能够将Ly6Chi MOMFs转换为Ly6Clo MOMFs,促进肝纤维化的消退[46]。
尽管肝巨噬细胞在肝纤维化发病机制中的作用机制和相关治疗策略已经取得了突破性进展,然而,通过巨噬细胞靶向肝纤维化疗法仍然存在局限性。需要解决的问题如下:肝巨噬细胞的这些表型其临床意义是什么,是否有可能对肝巨噬细胞进行基因改造以解决肝纤维化,如何达到只靶向致病表型而不破坏正常的生理表型?此外,大多数关于肝巨噬细胞的作用和潜在机制的研究都是在啮齿动物模型中进行的,由于啮齿动物和人类之间的肝巨噬细胞存有差异,这些发现与人类的相关性仍需要进一步研究。
Research progress on hepatic macrophages and liver fibrosis
-
摘要: 肝纤维化是由各种病因所致慢性肝损伤的修复反应,其持续进展可发展为肝硬化甚至肝细胞癌,最终导致肝功能衰竭。目前,肝纤维化尚无有效治疗方法。肝巨噬细胞在肝内炎症反应、纤维化的进展和消退方面发挥关键作用,已成为抗肝纤维化的重要治疗靶细胞。主要就肝巨噬细胞在肝纤维化过程中的作用进行综述,以期能够为肝纤维化治疗提供思路。Abstract: Liver fibrosis is a repair response to chronic liver injury caused by various etiologies. Its continuous progression can develop into liver cirrhosis or even hepatocellular carcinoma, eventually leading to liver failure. Currently, there is no effective treatment for liver fibrosis. Hepatic macrophages play a key role in intrahepatic inflammatory response, progression and resolution of fibrosis, and have emerged as an important therapeutic target for anti-hepatic fibrosis. The function of hepatic macrophages in the process of liver fibrosis was mainly reviewed and the mode of action of hepatic macrophages from various aspects was discussed to provide ideas for the treatment of liver fibrosis.
-
Key words:
- hepatic macrophages /
- liver fibrosis /
- phenotypic switch
-
西红花为名贵药材,来源于鸢尾科植物番红花Crocus sativus L.的干燥柱头。原产于地中海地区、希腊、小亚细亚和伊朗,后经西藏传入国内,故又名藏红花[1]。《本草纲目》中记载番红花“主治心忧郁积、气闷不散,活血,亦治惊悸”[2]。2020版《中国药典》描述西红花具有活血化瘀、凉血解毒、解郁安神的功效[3]。越来越多的现代药理研究表明,西红花具有抗肿瘤、抗血小板聚集与凋亡、抗心血管细胞凋亡、降血脂和降血糖等活性[4–6],在健康和医疗领域具有重要作用。
世界卫生组织国际癌症研究机构(IARC)发布的最新数据,2020年全球癌症新发患者病例数超过1 930万例,癌症死亡患者接近1 000万例[7]。天然活性成分是抗肿瘤药物研发的重要来源[8]。有研究表明,西红花中特有的西红花酸、西红花苷等具有抗肿瘤活性[9],已有学者在西红花治疗结直肠癌、乳腺癌等的抗肿瘤作用方面进行了相关研究[10-11],但其主要活性成分及抗肿瘤作用机制仍需进一步探索。
网络药理学[12]将系统生物学、生物信息学、计算生物学、网络科学和靶向药理学相结合,从系统层次和生物网络的整体角度探讨成分—靶标—通路的相互作用关系,为中药多靶点、多成分、系统性、整体性的作用机制研究提供了有力的技术支撑,从而指导新药研发和临床诊疗。因此,本研究应用网络药理学结合反向分子对接的方法,对西红花的抗肿瘤作用成分及靶点机制进行研究,为深入探索西红花抗肿瘤药效物质基础及作用机制提供参考。
1. 方法
1.1 西红花化学成分获取
利用TCMSP平台获取西红花化学成分,口服生物利用度(oral bioavailability,OB)和类药性(drug-likeness,DL) 是药物筛选的关键参数,一般设置OB≥30%和DL≥0.18的化学成分作为候选药效成分,并结合文献报道[13–15]补充4个西红花化学成分。
1.2 西红花活性成分和肿瘤疾病相关靶点整理
应用TCMSP平台和PharmMapper[16]工具获取西红花活性成分的作用靶点,并借助UniProt数据库将靶点转换为对应基因。以“tumor”、“cancer”为关键词,在GeneCards(https://www.genecards.org/)、OMIM数据库(https://www.omim.org/)和TTD数据库(http://db.idrblab.net/ttd/)进行检索。将得到的疾病靶点和药物靶点取交集,作为药物作用于疾病的预测靶点。
1.3 “成分-靶点”网络的构建
根据预测的西红花药效成分、交集靶点,使用Cytoscape 3.9.1软件建立“成分-靶点”的网络图。
1.4 蛋白质相互作用网络(protein-protein interaction,PPI)的构建与分析
将药物疾病交集靶点输入String数据库构建PPI网络进行初步筛选,再将PPI网络导入Cytoscape 3.9.1中,以半数degree为参考标准,选取关键靶点。
1.5 基因功能注释和富集通路分析
将筛选获得的37个核心靶点录入Metascape平台(http://metascape.org/gp/index.html),物种设置为人,选择Custom Analysis,设置P<0.01,进行基因本体(gene ontology,GO)功能富集分析及京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)通路富集分析。
1.6 分子对接
将筛选出的西红花主要活性成分通过PubChem下载SDF格式;利用RCSB PDB数据库下载关键蛋白靶点,优先选择有配体、结构相对完整的晶体结构,并采用AutoDock Tools对获取的PDB蛋白分子进行除水、加氢、计算电荷预处理;使用AutoDock Vina进行分子对接,计算结合能;选取最优构象,使用PyMOL软件做出3D结合模式图。
2. 结果
2.1 西红花化学成分获取
通过TCMSP获得70个西红花化学成分,设置OB≥30%且DL≥0.18进行筛选,再添加文献检索相关成分,共获得9个西红花活性成分,见表1。
表 1 西红花活性成分序号 化合物编号 化合物英文名 中文名 OB (%) DL 1 MOL001389 n-heptanal 庚醛 79.74 0.59 2 MOL001406 crocetin 西红花酸 35.3 0.26 3 MOL000354 isorhamnetin 异鼠李素 49.6 0.31 4 MOL000422 kaempferol 山柰酚 41.88 0.24 5 MOL000098 quercetin 槲皮素 46.43 0.28 6 MOL001405 crocin Ⅰ 西红花苷Ⅰ 2.54 0.12 7 MOL001407 crocin Ⅱ 西红花苷Ⅱ 1.65 0.21 8 MOL000720 safranal 藏红花醛 39.56 0.04 9 MOL001409 picrocrocin 苦番红花素 33.71 0.04 2.2 西红花活性成分和肿瘤疾病靶点
将TCMSP平台和PharmMapper获取结果进行整理,并借助UniProt数据库进行靶基因匹配,获得201个潜在靶点。以“tumor”和“cancer”为关键词,在GeneCards、OMIM和TTD数据库进行预测整理,剔除重复,筛选得到5896个潜在疾病靶点。将得到的疾病靶点和药物靶点取交集,共得到可作为药物作用于疾病的179个预测靶点。
2.3 “成分-靶点”网络的构建与分析
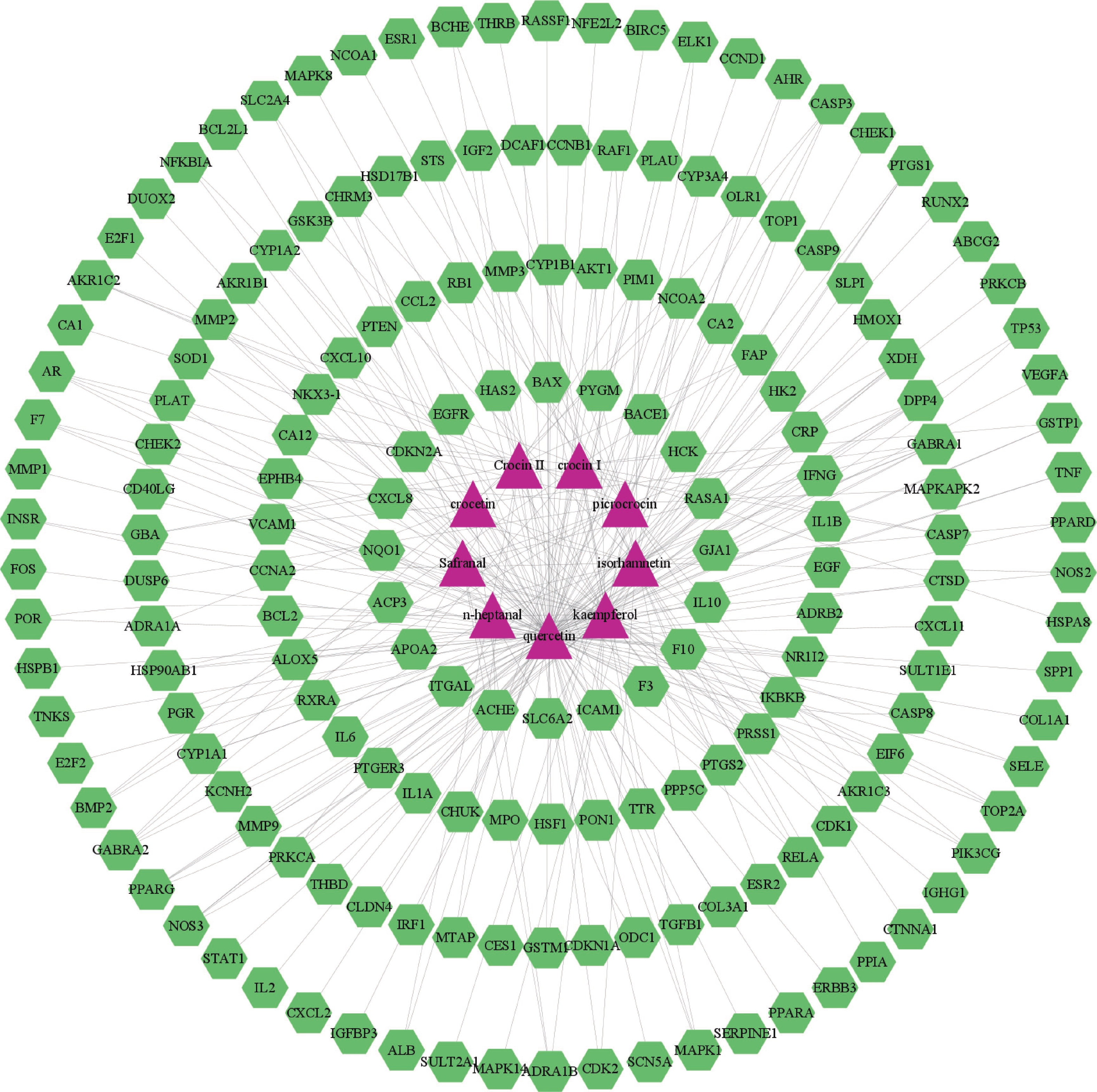
将西红花的9个活性成分与预测到的179个潜在靶点导入Cytoscape 3.9.1软件,构建“药物-活性成分-靶点”网络(图1),网络中绿色代表药物作用于疾病的靶点,蓝色代表西红花活性成分,全图包括189个节点、299条边,其中degree值排名靠前的活性成分为槲皮素、山柰酚、异鼠李素、苦番红花素和西红花苷Ⅰ,这些可能是西红花发挥抗肿瘤作用的潜在活性成分。
2.4 蛋白相互作用PPI的构建与分析
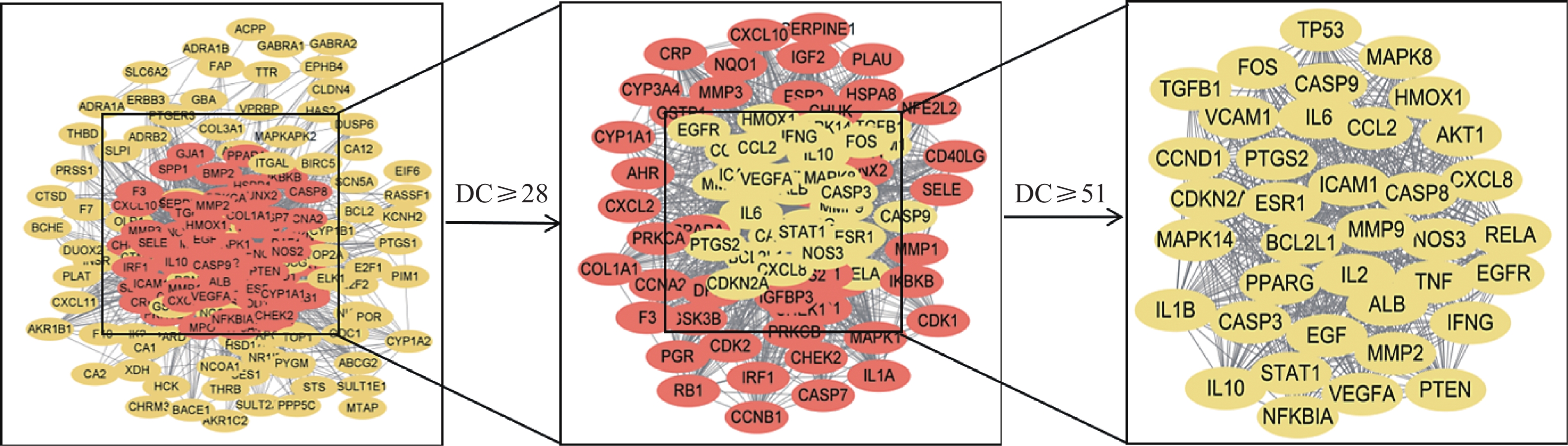
将疾病与活性成分的潜在靶点导入String数据库,采用Cytoscape 3.9.1软件绘制PPI网络图,依据degree值进行排序,以大于半数degree值为标准进行两次筛选,获取核心靶点37个(图2)。度值排名前5的靶点分别为EGF、MMP9、NFKBIA、IL-1B和IL-10,提示这些靶点可能是西红花发挥抗肿瘤作用的关键潜在靶点。
2.5 基因功能注释和富集通路分析
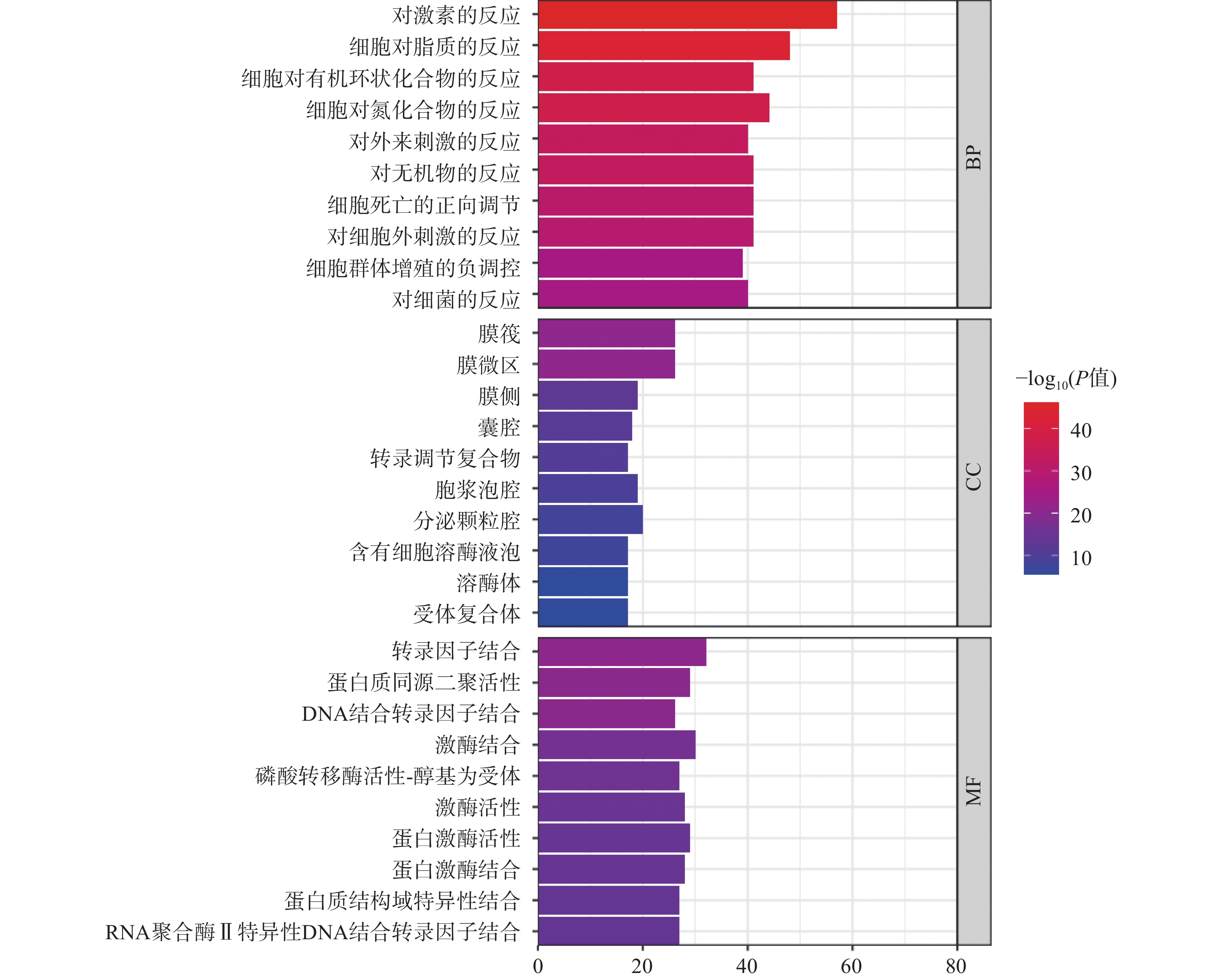
GO分析常用于注释基因和基因产物生物功能,分析包括生物过程(biological process,BP)、分子功能(molecular function,MF)和细胞组成(cellular component,CC)三部分。此次GO富集分析共得到BP富集结果193个、CC富集结果83个和MF富集结果123个,选取排名前10的条目绘制GO功能分析图(图3)。如图3所示,BP主要涉及对激素的反应、对脂质的反应、对异源刺激的反应等;CC主要涉及膜筏、膜微区、囊腔、细胞质囊泡腔等;MF主要涉及转录因子结合、DNA结合转录因子结合、RNA聚合酶Ⅱ特异性DNA结合转录因子结合等。通过比较发现,细胞生物过程富集的基因数较多,说明西红花可能主要通过调节生物过程发挥抗肿瘤作用。
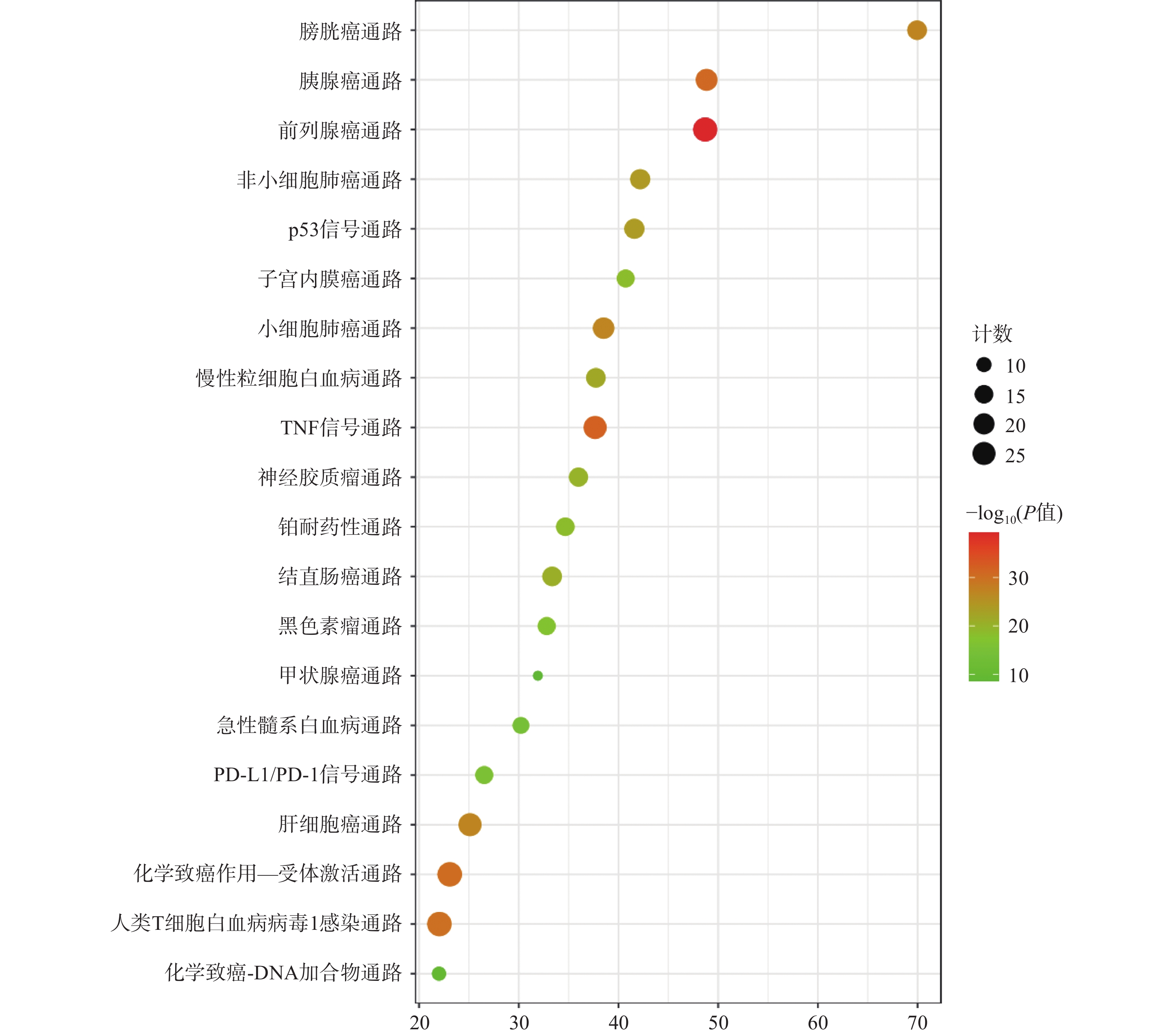
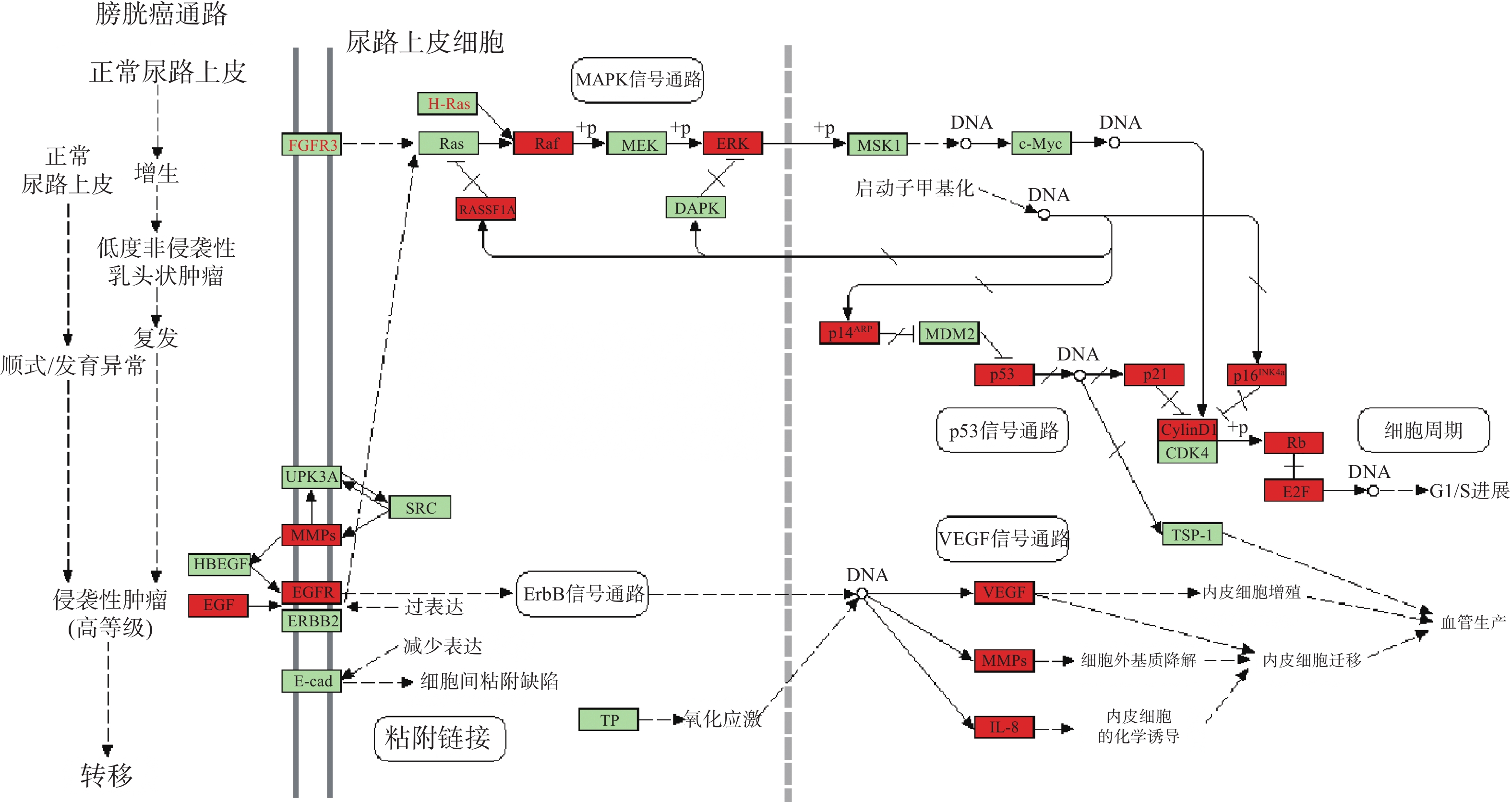
KEGG分析共富集到194条信号通路,其中34条癌症相关通路,并对前20条通路绘制气泡图(图4)。依据KEGG分析,西红花可能通过p53信号通路、TNF通路发挥抗肿瘤作用,可能对膀胱癌、胰腺癌、前列腺癌、非小细胞肺癌等肿瘤具有治疗作用,西红花靶点-通路相互作用网络见图5,红色三角形代表与肿瘤相关的信号通路,蓝色矩形代表关键靶点。其中,西红花通过膀胱癌信号通路调控EGF、MMPs、Raf、VEGF、ERK等基因发挥抗肿瘤作用(图6),红色矩形代表西红花可能干预的关键靶点。
2.6 分子对接
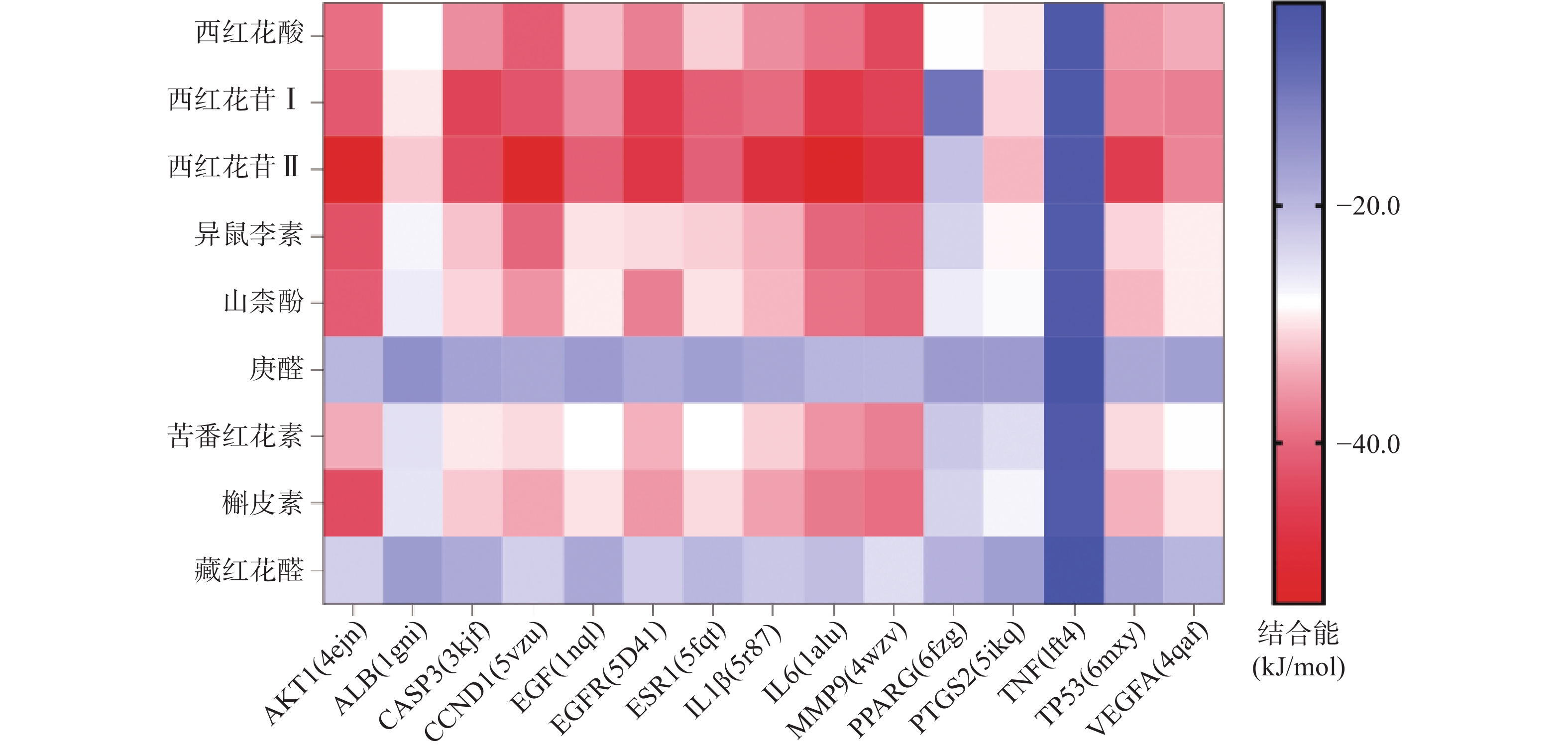
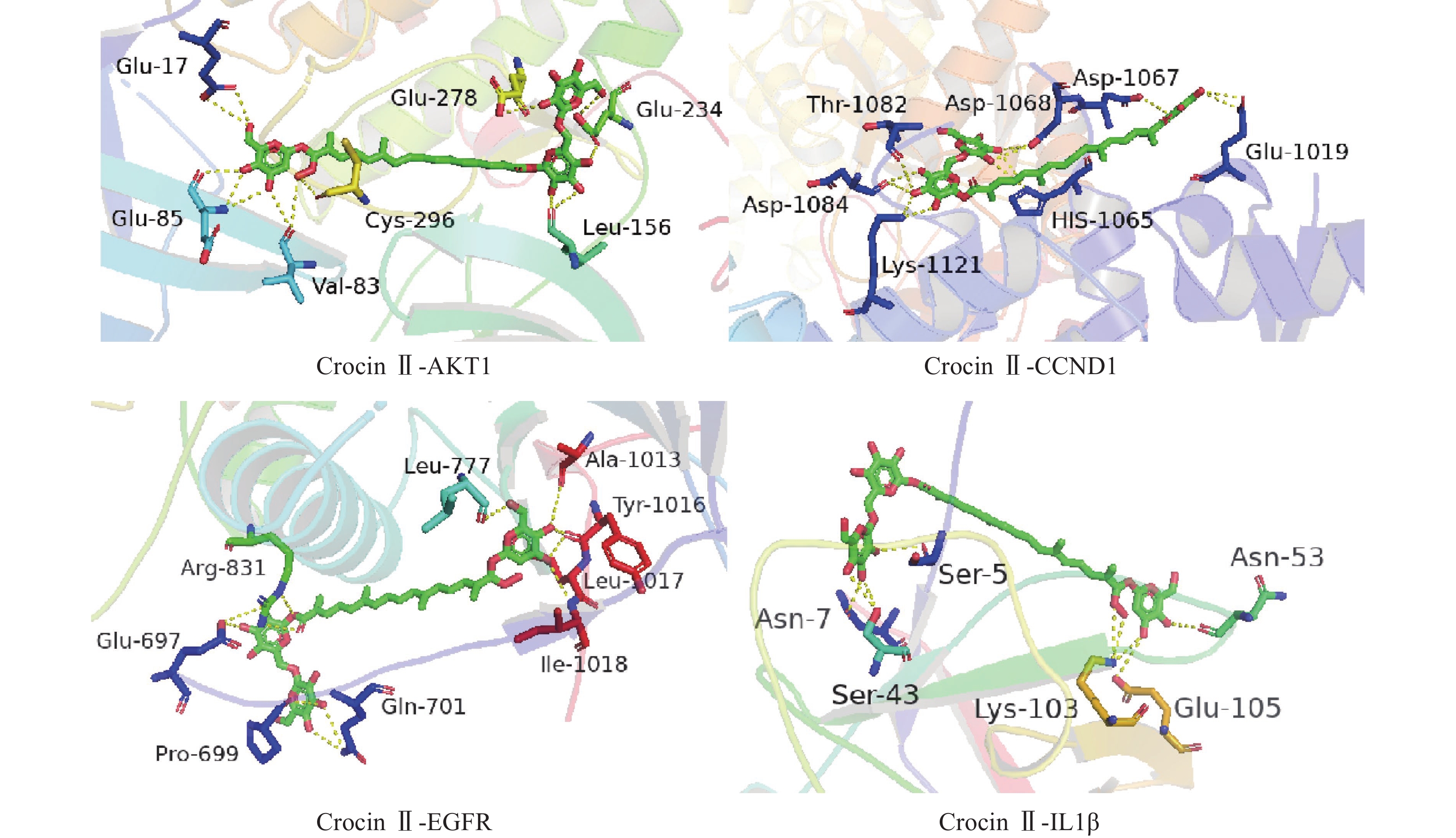
将前15个潜在核心靶点与西红花活性成分进行分子对接。结合能(affinity)<0表明配体分子能够与受体蛋白自发结合,结合能≤−17.78 kJ/mol表明配体与受体有一定的结合活性,结合能≤−20.92 kJ/mol 表明配体与受体有较好的结合活性,结合能≤−29.29 kJ/mol 表明配体与受体有强的结合活性[17],且结合能越低,表明对接的效果越好,结合的构象越稳定[18]。经AutoDock Vina对接,将得到的结合能数据使用热图展示(图7)。本研究结合自由能小于−20.92kJ/mol 的活性成分有102个,占75.6%;小于−29.29kJ/mol 的活性成分有73个,占54.1%,可见这些核心化合物与受体结合活性较高,结构相对稳定。选取结合能力最好的4个组合用Pymol软件进行可视化处理(图8)。
3. 讨论
肿瘤的发生和发展是多基因、多步骤的结果。中药多成分、多靶点的特点使其在肿瘤治疗方面有独特的优势。大量的临床实践表明,中药在治疗肿瘤中能够改善症状、提高患者生存质量、延长生存期等,有着其他治疗药物及手段不可替代的作用[19-20]。以中药黄芪为例,不仅可以通过Wnt5/β-catenin信号通路抑制肿瘤生长[21],同时具有通过PD-L1下调诱导耐药黑色素瘤的干性抑制和化疗敏感性增强的作用,可以减少化疗药物用量[22],还能充当免疫佐剂,提高患者免疫力,改善生存质量[23]。网络药理学最大的优势在于可以运用系统生物学的分析,为中药多成分、多靶点、多通路的机制研究提供有力的技术支撑[12],其分析理念和技术路径又与中医药治疗疾病的整体观相契合,已用于多种中药和中药复方作用机制的研究,如灯盏细辛、半枝莲等中药和茵陈蒿汤、桃红四物汤等中药复方,利用网络药理学的方法得到治疗机制的详细阐述和证明[24–27],为中药药理作用机制的探索提供了很好的参考。
本研究发现西红花中多种成分,如西红花酸、西红花苷等可与IL-6、AKT1、CCND1、IL-1β、MMP9、EGFR、TP53靶点产生适度结合,提示这些靶点可能是西红花中活性成分发挥抗肿瘤作用的关键靶点。研究发现,AKT1是PI3K-AKT-mTOR信号通路中的重要靶点,被磷酸化激活后可以促进细胞的增殖与存活,与肿瘤细胞的生长密切相关[28]。多项研究表明,通过抑制AKT1可以治疗肺癌、结肠癌、卵巢癌等多种实体癌[29]。EGFR与一些全球发病率和致死率高的癌症发病机制直接或间接相关,包括肺癌、乳腺癌和结直肠癌等[30]。当EGFR过度表达时,细胞表面会出现过量的受体,诱导正常的细胞转化为癌细胞,并为癌细胞持续生存提供条件[31]。CCND1是细胞周期家族的一员[32],公认的原癌基因,在甲状旁腺瘤、乳腺癌、肝癌及食管、肺、头颈部鳞状细胞癌的发生、发展过程中均起着重要作用[33-34]。西红花苷是由西红花酸和龙胆二糖或葡萄糖结合形成的二萜苷类化合物,西红花苷Ⅰ和西红花苷Ⅱ的差别在于分子中糖苷基数目的多少 [35]。西红花酸已具有抗肿瘤作用,以其为苷元形成的西红花苷同样也表现出较好的抗肿瘤活性,其中西红花苷Ⅱ的表现最好。西红花苷可以通过P53途径下调细胞周期蛋白d1和p21的表达,诱导细胞凋亡和细胞周期停滞,从而抑制肿瘤生长[36]。分子对接的结果与GO富集分析、KEGG通路富集分析结果一致,验证了网络药理学分析结果的正确性。
Buyun等学者在肝癌Hep3B和HepG2细胞中使用西红花苷抑制了IL-6对STAT3以及细胞周期蛋白D1的激活,验证了西红花对肝癌细胞的抗增殖,凋亡和阻断入侵作用[37]。在转移性乳腺癌的研究中,Ali等研究人员在体内和体外实验中均证明了西红花苷可以通过VEGF和MMP9下调发挥抗肿瘤作用,而且对乳腺癌的转移扩散有较好的抑制作用[38]。这些研究成果在一定程度上验证了利用网络药理学探究发现的西红花抗肿瘤作用机制的可行性。
综上所述,本研究利用网络药理学结合分子对接技术,探究西红花抗肿瘤作用的活性成分、作用靶点及信号通路。发现西红花抗肿瘤的作用具有多成分、多靶点、多通路、多机制的特点,其中以西红花苷为代表的西红花特有化学成分显示出了良好的抗肿瘤活性,可以在多条肿瘤发生通路中发挥作用,为西红花治疗肿瘤的深入研究提供了理论基础。但这些结果受限于各个数据库信息的片面性,而且只关注了成分,没有考量到成分的含量及其之间是否存在相互作用,预测的结果存在一定的片面性和局限性,需要进一步进行体内、外实验验证。
-
[1] KHATUN M, RAY R B. Mechanisms underlying hepatitis C virus-associated hepatic fibrosis[J]. Cells, 2019, 8(10):1249. doi: 10.3390/cells8101249 [2] TACKE F. Targeting hepatic macrophages to treat liver diseases[J]. Hepatol, 2017, 66(6):1300-1312. doi: 10.1016/j.jhep.2017.02.026 [3] PRADERE J P, KLUWE J, DE MINICIS S, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice[J]. Hepatology, 2013, 58(4):1461-73. doi: 10.1002/hep.26429 [4] LI J, ZHAO Y P, TIAN Z. Roles of hepatic stellate cells in acute liver failure: From the perspective of inflammation and fibrosis[J]. World J Hepatol, 2019, 11(5):412-420. doi: 10.4254/wjh.v11.i5.412 [5] WEN Y K, LAMBRECHT J, JU C, et al. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities[J]. Cell Mol Immunol, 2021, 18(1):45-56. doi: 10.1038/s41423-020-00558-8 [6] KIAGIADAKI F, KAMPA M, VOUMVOURAKI A, et al. Activin-A causes Hepatic stellate cell activation via the induction of TNFα and TGFβ in Kupffer cells[J]. Biochim Biophys Acta Mol Basis Dis, 2018, 1864(3):891-899. doi: 10.1016/j.bbadis.2017.12.031 [7] SASAKI R, DEVHARE P B, STEELE R, et al. Hepatitis C virus-induced CCL5 secretion from macrophages activates hepa-tic stellate cells[J]. Hepatology, 2017, 66(3):746-757. doi: 10.1002/hep.29170 [8] NIELSEN S R, QUARANTA V, LINFORD A, et al. Macro-phage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis[J]. Nat Cell Biol, 2016, 18(5):549-560. doi: 10.1038/ncb3340 [9] GE X D, ARRIAZU E, MAGDALENO F, et al. High mobility group box-1 drives fibrosis progression signaling via the receptor for advanced glycation end products in mice[J]. Hepatology, 2018, 68(6):2380-2404. doi: 10.1002/hep.30093 [10] SHIRABE K, BEKKI Y, GANTUMUR D, et al. Mac-2 binding protein glycan isomer (M2BPGi) is a new serum biomarker for assessing liver fibrosis: more than a biomarker of liver fibrosis[J]. J Gastroenterol, 2018, 53(7):819-826. doi: 10.1007/s00535-017-1425-z [11] WANG C, MA C, GONG L H, et al. Macrophage polarization and its role in liver disease[J]. Front Immunol, 2021, 12:803037. doi: 10.3389/fimmu.2021.803037 [12] SINGANAYAGAM A, TRIANTAFYLLOU E. Macrophages in chronic liver failure: diversity, plasticity and therapeutic targeting[J]. Front Immunol, 2021, 12:661182. doi: 10.3389/fimmu.2021.661182 [13] XU J J, ZHU L, LI H D, et al. DNMT3a-mediated methylation of PSTPIP2 enhances inflammation in alcohol-induced liver injury via regulating STAT1 and NF-κB pathway[J]. Pharmacol Res, 2022, 177:106125. doi: 10.1016/j.phrs.2022.106125 [14] SU S B, QIN S Y, XIAN X L, et al. Interleukin-22 regulating Kupffer cell polarization through STAT3/Erk/Akt crosstalk pathways to extenuate liver fibrosis[J]. Life Sci, 2021, 264:118677. doi: 10.1016/j.lfs.2020.118677 [15] TOSELLO-TRAMPONT A C, KRUEGER P, NARAYANAN S, et al. NKp46+ natural killer cells attenuate metabolism-induced hepatic fibrosis by regulating macrophage activation in mice[J]. Hepatology, 2016, 63(3):799-812. doi: 10.1002/hep.28389 [16] CALVENTE C J, TAMEDA M, JOHNSON C D, et al. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223[J]. J Clin Investig, 2019, 129(10):4091-4109. doi: 10.1172/JCI122258 [17] SHE S P, WU X N, ZHENG D F, et al. PSMP/MSMP promotes hepatic fibrosis through CCR2 and represents a novel therapeutic target[J]. J Hepatol, 2020, 72(3):506-518. doi: 10.1016/j.jhep.2019.09.033 [18] SUN H F, FENG J G, TANG L L. Function of TREM1 and TREM2 in liver-related diseases[J]. Cells, 2020, 9(12):2626. doi: 10.3390/cells9122626 [19] KOHYAMA M, MATSUOKA S, SHIDA K, et al. Monocyte infiltration into obese and fibrilized tissues is regulated by PILRα[J]. Eur J Immunol, 2016, 46(5):1214-1223. doi: 10.1002/eji.201545897 [20] RAMACHANDRAN P, DOBIE R, WILSON-KANAMORI J R, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level[J]. Nature, 2019, 575(7783):512-518. doi: 10.1038/s41586-019-1631-3 [21] ZHOU H M, ZHONG W Z, RAO Z, et al. Defective mitophagy in aged macrophages promotes mitochondrial DNA cytosolic leakage to activate STING signaling during liver sterile inflammation [J]. Aging Cell, 2022, 21(6):e13622. [22] YU Y S, LIU Y, AN W S, et al. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis[J]. J Clin Invest, 2019, 129(2):546-555. [23] HU Q, LYON C J, FLETCHER J K, et al. Extracellular vesicle activities regulating macrophage- and tissue-mediated injury and repair responses[J]. Acta Pharm Sin B, 2021, 11(6):1493-1512. doi: 10.1016/j.apsb.2020.12.014 [24] ROSSO C, KAZANKOV K, YOUNES R, et al. Crosstalk between adipose tissue insulin resistance and liver macrophages in non-alcoholic fatty liver disease[J]. J Hepatol, 2019, 71(5):1012-1021. doi: 10.1016/j.jhep.2019.06.031 [25] LESLIE J, MACIA M G, LULI S, et al. C-rel orchestrates energy-dependent epithelial and macrophage reprogramming in fibrosis[J]. Nat Metab, 2020, 2(11):1350-1367. doi: 10.1038/s42255-020-00306-2 [26] KANAMORI Y, TANAKA M, ITOH M, et al. Iron-rich Kupffer cells exhibit phenotypic changes during the development of liver fibrosis in NASH[J]. iScience, 2021, 24(2):102032. doi: 10.1016/j.isci.2020.102032 [27] XU F, GUO M M, HUANG W, et al. Annexin A5 regulates hepatic macrophage polarization via directly targeting PKM2 and ameliorates NASH[J]. Redox Biol, 2020, 36:101634. doi: 10.1016/j.redox.2020.101634 [28] SUN K, XU L Y, JING Y Y, et al. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-κB-IL1α/β-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis[J]. Cancer Lett, 2017, 388:198-207. doi: 10.1016/j.canlet.2016.12.004 [29] CHENG D, CHAI J, WANG H W, et al. Hepatic macrophages: key players in the development and progression of liver fibrosis[J]. Liver Int, 2021, 41(10):2279-2294. doi: 10.1111/liv.14940 [30] ZHU J F, ZHANG W W, ZHANG L N, et al. IL-7 suppresses macrophage autophagy and promotes liver pathology in Schistosoma japonicum-infected mice[J]. J Cell Mol Med, 2018, 22(7):3353-3363. doi: 10.1111/jcmm.13610 [31] WAN J H, WEISS E, BEN MKADDEM S, et al. LC3-associated phagocytosis protects against inflammation and liver fibrosis via immunoreceptor inhibitory signaling[J]. Sci Transl Med, 2020, 12(539):eaaw8523. doi: 10.1126/scitranslmed.aaw8523 [32] AN P, WEI L L, ZHAO S S, et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis[J]. Nat Commun, 2020, 11:2362. doi: 10.1038/s41467-020-16092-0 [33] LI L A, WEI W, LI Z Z, et al. The spleen promotes the secretion of CCL2 and supports an M1 dominant phenotype in hepatic macrophages during liver fibrosis[J]. Cell Physiol Biochem, 2018, 51(2):557-574. doi: 10.1159/000495276 [34] ZHAO X T, WANG J, DENG Y, et al. Quercetin as a protective agent for liver diseases: a comprehensive descriptive review of the molecular mechanism[J]. Phytother Res, 2021, 35(9):4727-4747. doi: 10.1002/ptr.7104 [35] LIU P, LI H, GONG J S, et al. Chitooligosaccharides alleviate hepatic fibrosis by regulating the polarization of M1 and M2 macrophages[J]. Food Funct, 2022, 13(2):753-768. doi: 10.1039/D1FO03768D [36] ZHENG Z Q, WANG H A, LI L A, et al. Splenectomy enhances the Ly6Clow phenotype in hepatic macrophages by activating the ERK1/2 pathway during liver fibrosis[J]. Int Immunopharmacol, 2020, 86:106762. doi: 10.1016/j.intimp.2020.106762 [37] ZHAO X G, CHEN G M, LIU Y, et al. Curcumin reduces Ly6Chi monocyte infiltration to protect against liver fibrosis by inhibiting Kupffer cells activation to reduce chemokines secretion[J]. Biomed Pharmacother, 2018, 106:868-878. doi: 10.1016/j.biopha.2018.07.028 [38] AMBADE A, LOWE P, KODYS K, et al. Pharmacological inhibition of CCR2/5 signaling prevents and reverses alcohol-induced liver damage, steatosis, and inflammation in mice[J]. Hepatology, 2019, 69(3):1105-1121. doi: 10.1002/hep.30249 [39] AIDA H, DINA C, WAN J H, et al. Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver[J]. Gut, 2019, 68(3):522-532. doi: 10.1136/gutjnl-2018-316137 [40] FRIEDMAN S L, RATZIU V, HARRISON S A, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis[J]. Hepatology, 2018, 67(5):1754-1767. doi: 10.1002/hep.29477 [41] MORONI F, DWYER B J, GRAHAM C, et al. Safety profile of autologous macrophage therapy for liver cirrhosis[J]. Nat Med, 2019, 25(10):1560-1565. doi: 10.1038/s41591-019-0599-8 [42] WEISKIRCHEN R, TACKE F. Liver fibrosis: from pathogenesis to novel therapies[J]. 2016, Dig Dis, 34(4): 410-422. [43] LOOMBA R, LAWITZ E, MANTRY P S, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial[J]. Hepatology, 2018, 67(2):549-559. doi: 10.1002/hep.29514 [44] RAMACHANDRAN P, PELLICORO A, VERNON M A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis[J]. Proc Natl Acad Sci U S A. 2012, 109(46): E3186-3195. [45] KOYAMA Y, BRENNER D A. Liver inflammation and fibrosis[J]. J Clin Invest, 2017, 127(1):55-64. doi: 10.1172/JCI88881 [46] BAECK C, WEI X, BARTENECK M, et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice[J]. Hepatology, 2014, 59(3):1060-1072. doi: 10.1002/hep.26783 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 6912
- HTML全文浏览量: 2819
- PDF下载量: 82
- 被引次数: 0
