

 下载:
下载:

-
钆特酸葡胺注射液是一种临床上常用的含钆增强磁共振对比剂。钆对比剂按照钆离子结合配体的结构不同,可将其分为线性类和大环类两类,钆特酸葡胺注射液中钆属于大环类钆对比剂。钆对比剂具有很强的亲水性,理论上不能进入细胞内,且无特殊靶器官。通过静注后短时间内分布到全身血液系统,并很快达到平衡期,可在1 d内以原型的形式全部排出体外,理论上在人体内无残留。近年来研究表明,反复使用钆对比剂的患者,其脑部会出现不同程度的钆对比剂沉积现象,并且其脑深部核团的MRI扫描信号也会随之发生改变[1-3]。同时,使用不同类型的钆对比剂所造成的钆对比剂脑沉积的程度也有所不同[4-5]。曾经反复使用过线状钆对比剂的患者,可在其头颅MR扫描中观察到明显的信号改变。而曾经多次使用过大环状钆对比剂的患者,在脑中却未观察到明显的钆对比剂沉积现象[6]。在此基础上,美国食品药品监督管理局发布通告,要求医疗人员重新评估使用钆对比剂的条件[7]。同时,欧洲药品管理局也停止了对几种线性钆对比剂的市场授权[8]。2017年,我国食品药品监督管理局也发布通告提示关注含钆对比剂反复使用引起脑部钆沉积的风险[9-10]。研究表明,钆对比剂中游离Gd3+的含量与钆对比剂的脑沉积现象密切相关[4-5],但目前无论在体内或者体外环境中定量测定钆对比剂中游离Gd3+含量的方法尚不成熟。因此,笔者通过反复试验并查阅相关资料,建立了一种新的体外环境测定钆对比剂中游离Gd3+含量的方法。
-
AKTA蛋白纯化仪、自动进样器(美国GE公司);ICP-MS 8800(Agilent technologies);分离柱(1-ml Chelating Sepharose column GE)。
-
钆特酸葡胺注射液0.5 mol/L,含钆特酸葡胺377 g/L。(批号:170923BC、170924BC、170925BC,江苏恒瑞医药股份有限公司);Gd标准对照品溶液(标准值1 000 mg/L,批号:GSB 04-1780-2004,国家有色金属及电子材料分析测试中心);六水合硝酸钆(浓度99.99%);去离子水(18.2 MΩ,自制);内标溶液:20 mg/L的6Li、45Sc、89Y、115In、209Bi混合溶液(2%硝酸介质,Accu Standard,USA)。其他试剂均为分析纯。
-
分离柱为金属螯合柱(1-ml Chelating Sepharose column),常温下,A液为10 mmol/L的双(2-羟乙基)氨基(三羟甲基)甲烷(BIS-TRIS)溶液,B液为50 mmol/L的EDTA溶液。流速为1 ml/min,平衡系统后,进样500 μl,先用100%的A液冲洗10 min(10倍柱体积),再用100%的B液冲洗20 min(20倍柱体积),得到分离后的样品。
-
载气为氩气,射频功率1 350 W,冷却气流量:13 L/min;辅助气流量:0.80 L/min;雾化器氩气流量:0.88 L/min。采用内标法做定量分析,将“2.1.1”项分离得到的样品稀释10倍后,用过蠕动泵进样,样品提升速度为0.2 r/min,时间45 s,样品平衡速度0.1 r/min,时间35 s。ICP-MS的相对分子质量为157。
-
精密量取标准钆对照品1 ml,置50 ml容量瓶中,加0.5%的稀硝酸稀释至刻度,摇匀即得20 mg/L标准对照品储备液。
-
精密量取钆特酸葡胺注射液1 ml,置于50 ml量瓶中,加去离子水稀释至刻度,摇匀,即得10 mmol/L样品储备液。
-
取六水合硝酸钆(99.99%)0.45 g,精密称量,置于1 000 ml容量瓶中,加去离子水稀释至刻度,摇匀得到1 mmol/L的中性对照品储备液。
-
精密吸取“2.2.3”项下对照品储备液1.0 ml,置于10 ml量瓶中,再精密吸取“2.2.2”供试品储备液1 ml,用去离子水稀释至刻度,摇匀。得到1 mmol/L供试品(Gd配合物)+0.1 mmol/L中性对照品混合溶液(Gd3+)。
-
精密吸取“2.2.3”中性对照品储备液1.0 ml,置于10 ml量瓶中,用去离子水稀释至刻度,摇匀,得到0.1 mmol/L的中性对照品溶液(Gd3+)。精密吸取1 ml上述稀释后的中性对照品储备液得到样本1。精密吸取“2.2.2”供试品储备液1.0 ml,置于10 ml量瓶中,用去离子水稀释至刻度,摇匀,得到1 mmol/L的供试品溶液(Gd配合物)。精密吸取1 ml上述稀释后的供试品储备液得到样本2。取“2.2.4”项下溶液1 ml,作为样本3。将样本1、2、3分别稀释10倍后,照“2.1.1”项下分离条件分离供试品,并间隔2 min收取1管分离后的供试品。得到3批、共45管分离后的供试品。将分离后的供试品按照“2.1.2”项下ICP-MS条件测量其浓度变化,结果见图1。混合对照品质谱中,有两个保留时间的质谱峰,分别对应对照品质谱和供试品质谱相应的位置上的质谱峰,而两个质谱峰之间无干扰。
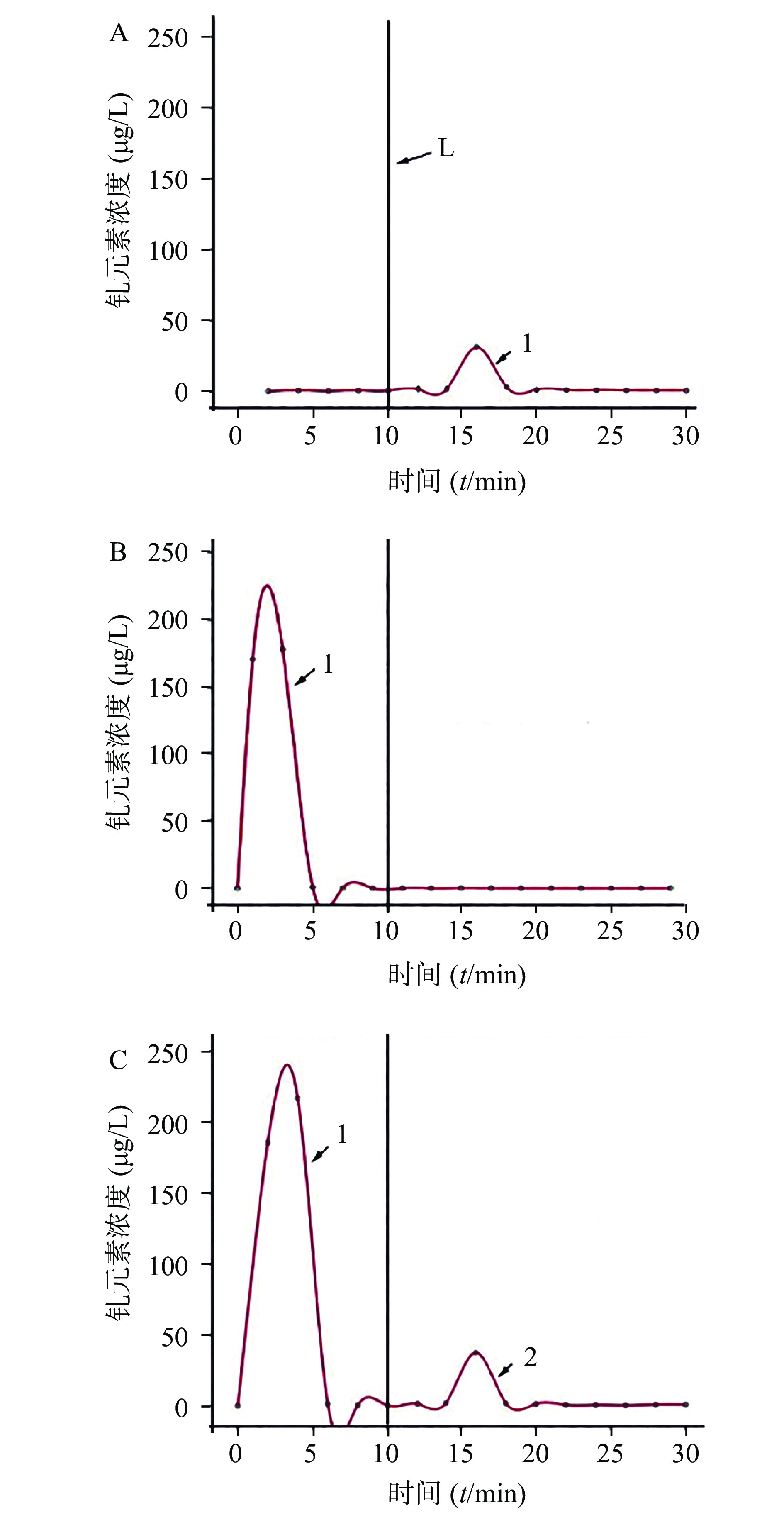
图 1 方法专属性考察曲线
-
精密吸取“2.2.1”项下标准对照品储备液0.5、1.25、2.5 ml,加入到100 ml容量瓶中。用0.5%的稀硝酸稀释至刻度线,摇匀。得到理论浓度为100、250、500 μg/L的标准对照品溶液。再分别取浓度为500和250 μg/L的溶液5 ml置于50 ml容量瓶中,用0.5%的稀硝酸稀释至刻度线,摇匀。得到理论浓度为25、50、100、250、500 μg/L的标准对照品溶液,按上述ICP-MS条件进样测定,以对照品浓度为横坐标(X,μg/L),对照品峰面积为纵坐标(Y),绘制标准曲线。回归方程:Y=0.001 635X+0.000 000E+000(经过原点),r=1.000。结果表明Gd元素浓度在0~500 μg/L浓度范围内线性关系良好。
-
精密吸取“2.2.1”项下标准对照品储备液0.2 ml,用0.5%硝酸稀释至40 ml,摇匀。按上述ICP-MS条件进样测定,连续操作5次,测得钆浓度。结果表明,观察峰面积的RSD <10%(n=5),表明仪器精密度良好。结果见表1。
表 1 钆特酸葡胺注射液精密度试验结果
样品编号 测得Gd浓度(μg/L) 峰面积RSD(%) 1 94.92 2.69 2 98.24 1.69 3 95.43 2.46 4 104.91 4.08 5 90.56 3.13 -
取“2.2.4”项下同一供试品溶液适量,稀释10倍后,按“2.1.1”项下条件进行分离,分别取分离后前10 min和后20 min的溶液,编号为a液和b液。分别精密量取a液、b液各5份,于室温下放置0、2、4、6、12 h,按“2.1.2”项下的条件进样测定。结果显示,a液的RSD为1.826%(n=5),RSD<2%表明分离后的钆配合物12 h内稳定性良好。b液的RSD为1.911%(n=5),表明分离后的钆与EDTA螯合物在12 h内稳定性良好(表2)。
表 2 钆特酸葡胺注射液稳定性试验结果(n=5)
室温下放置时间(t/h) 测得Gd浓度(μg/L) a液 b液 0 81.18 4.08 2 83.14 4.17 4 81.05 4.11 6 80.41 3.97 12 79.05 4.02 -
取同一批样品(批号:170923BC),精密量取适量,按照“2.2.4”项下方法配成混合溶液适量,稀释10倍后,按“2.1.1”项下条件进行分离,分别取分离后前10 min和后20 min的溶液,编号为a液和b液。分别按“1.1.2”项下条件进样测定。计算混合溶液中Gd3+的实际含量。同法操作重复5次。结果表明,混合溶液中Gd3+的实测含量RSD为1.998%(n=5),RSD<2%(n=5)表明本方法重复性良好,结果见表3。
表 3 钆特酸葡胺注射液重复性试验结果(n=5,mmol/L)
样品编号 Gd3+的理论含量 Gd3+的实测含量 1 0.1 0.104 2 0.1 0.106 3 0.1 0.105 4 0.1 0.101 5 0.1 0.102 -
用空白对照溶液(去离子水),按“2.1.2”项下ICP-MS条件进样测定,连续操作8次,分别测得钆浓度。计算其平均浓度,检测限=测得空白对照的平均浓度×3,测得结果为0.223 μg/L。
-
用空白对照溶液(0.5%的硝酸),按“2.1.2”项下ICP-MS条件进样测定,连续操作8次,分别测得钆浓度。计算其平均浓度,定量限=测得空白对照的平均浓度×10,测得结果为:0.7443 μg/L。
-
精密量取已知含量的钆特酸葡胺注射液(批号:170923BC)4份,按照“2.2.2”方法配得10 mmol/L的样品溶液,精密量取1 ml样品溶液置10 ml容量瓶中,用0.5%的硝酸定容,摇匀,得到1 mmol/L的样品溶液。精密量取1 ml稀释后的样品,用0.5%的硝酸再稀释到30 ml。得到5.23 mg/L的样品溶液,称其为①液。精密量取1 ml对照品溶液(1 g/L)置50 ml容量瓶中,用0.5%的硝酸定容,摇匀。同样精密量取5 ml稀释后的对照品溶液,用0.5%的硝酸稀释至20 ml,得到5 mg/L的对照品溶液,称为②液。精密量取2 ml①液置50 ml容量瓶中,用0.5%的硝酸定容,摇匀,得到样品液A。分别精密量取1 ml①液和1 ml②液置50 ml容量瓶中,用0.5%的硝酸定容,摇匀,得到样品液B。得到的两份溶液分别按“2.1.2”项下条件,进样测定并计算加样回收率,反复4次上述步骤。加样回收率=(加样试样测定值-加样测定值)/加样量×100%。计算平均加样回收率,结果见表4。
表 4 加样回收率试验结果(n=4)
样品理论钆含量
(m/mg)标准品理论钆含量
(m/mg)样品钆含量
(m/mg)混合溶液钆含量
(m/ng)加样回收率
(%)平均加样回收率
(%)RSD
(%)104.7 100 209.23 197.74 93.13 93.19 1.9 104.7 100 215.99 199.24 91.24 104.7 100 206.47 196.02 92.78 104.7 100 220.80 206.01 95.61 -
取3批精密量取已知含量的钆特酸葡胺注射液,按“2.2.2”项下方法制备供试品溶液,按“2.1”项下条件测定,通过回归方程计算,结果见表5。
表 5 钆特酸葡胺注射液Gd3+含量测定结果(n=3)
批号 含量(mg/L) RSD(%) 20160901 2.63 1.53 20160902 2.60 1.15 20160903 2.64 1.36 -
文献[11]报道,采用HPLC-ICP-MS联用,以硝酸作为洗脱液的方法测定钆特酸葡胺注射液中Gd3+的含量。经查阅相关资料,其选用的金属螯合柱实际为低压柱,无法承受高效液相的压力,并且硝酸的洗脱效率较EDTA要低得多,还具有腐蚀性,对于ICP-MS仪器有一定的损害,因此,该文献所报道方法无法验证。根据配位化学原理,EDTA对Gd3+有很强的螯合性,能快速形成稳定配合物,故笔者选用EDTA作为洗脱流动相,结果表明,此法可迅速将螯合柱上的Gd3+完全洗脱。因为自然界中Gd元素含量稀少,所以本实验运用ICP-MS时受外界干扰小,实验结果可靠。
本实验建立的测定钆特酸葡胺注射液中Gd3+含量的方法操作简便、结果准确、重现性好。研究表明不同批次钆特酸葡胺注射液样品中Gd3+含量变化较小,质量稳定可控。综上所述,此方法可作为测定钆特酸葡胺注射液中Gd3+含量的方法。
Determination of the content of Gd3+ in gadoteric acid meglumine salt injection by ICP-MS method
-
摘要:
目的 建立测定钆特酸葡胺注射液中Gd3+含量的方法。 方法 电感耦合等离子体质谱(ICP-MS)法。分离柱为金属螯合柱(1-ml Chelating Sepharose column),柱温为常温,流速为1 ml/min,进样量为0.5 ml。ICP-MS的相对分子质量为157(Gd的相对分子质量为157),载气为氩气。 结果 回归方程为:Y=0.001 635X+0.000 000E+000(经过原点),r=1.000。线性范围为0~500 μg/L;仪器精密度、稳定性、重复性、加样回收率试验结果均符合要求。 结论 该方法操作简便、结果准确、重复性好,可用于测定钆特酸葡胺注射液中Gd3+的含量。 -
关键词:
- 电感耦合等离子体质谱法 /
- 钆特酸葡胺注射液 /
- Gd3+ /
- 含量测定
Abstract:Objective To establish a method for determining the content of Gd3+ in gadoteric acid meglumine salt injection. Methods ICP-MS was used. The separation column was a metal chelate column (1-ml Chelating Sepharose column), column temperature was normal temperature. Flow rate was 1 ml/min. Injection volume was 500 μl. Atoms were measured by ICP-MS with a molecular weight of 157 (The molecular weight of Gd was 157). The carrier gas was argon. Results The linear range of Gd3+ mass concentration was 0-500 ng/ml (r=1.000); The precision, stability and repeatability of the sample recovery test were all in accordance with the requirements. Conclusion The method was simple in operation, accurate in results and good in repeatability, which could be used to determine the content of Gd3+ in gadoteric acid meglumine salt injection. -
Key words:
- ICP-MS /
- gadoteric acid meglumine salt injection /
- Gd3+ /
- content determination
-
表 1 钆特酸葡胺注射液精密度试验结果
样品编号 测得Gd浓度(μg/L) 峰面积RSD(%) 1 94.92 2.69 2 98.24 1.69 3 95.43 2.46 4 104.91 4.08 5 90.56 3.13  下载: 导出CSV
下载: 导出CSV
表 2 钆特酸葡胺注射液稳定性试验结果(n=5)
室温下放置时间(t/h) 测得Gd浓度(μg/L) a液 b液 0 81.18 4.08 2 83.14 4.17 4 81.05 4.11 6 80.41 3.97 12 79.05 4.02
下载: 导出CSV
表 3 钆特酸葡胺注射液重复性试验结果(n=5,mmol/L)
样品编号 Gd3+的理论含量 Gd3+的实测含量 1 0.1 0.104 2 0.1 0.106 3 0.1 0.105 4 0.1 0.101 5 0.1 0.102
下载: 导出CSV
表 4 加样回收率试验结果(n=4)
样品理论钆含量
(m/mg)标准品理论钆含量
(m/mg)样品钆含量
(m/mg)混合溶液钆含量
(m/ng)加样回收率
(%)平均加样回收率
(%)RSD
(%)104.7 100 209.23 197.74 93.13 93.19 1.9 104.7 100 215.99 199.24 91.24 104.7 100 206.47 196.02 92.78 104.7 100 220.80 206.01 95.61
下载: 导出CSV
表 5 钆特酸葡胺注射液Gd3+含量测定结果(n=3)
批号 含量(mg/L) RSD(%) 20160901 2.63 1.53 20160902 2.60 1.15 20160903 2.64 1.36
下载: 导出CSV
-
[1] KANDA T, ISHⅡ K, KAWAGUCHI H, et al. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material[J]. Radiology,2014,270(3):834-841. doi: 10.1148/radiol.13131669 [2] ROBERTS D R, HOLDEN K R. Progressive increase of T1 signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images in the pediatric brain exposed to multiple doses of gadolinium contrast[J]. Brain Dev,2016,38(3):331-336. doi: 10.1016/j.braindev.2015.08.009 [3] RADBRUCH A, WEBERLING L D, KIESLICH P J, et al. Gadolinium retention in the dentate nucleus and globus pallidus is dependent on the class of contrast agent[J]. Radiology,2015,275(3):783-791. doi: 10.1148/radiol.2015150337 [4] ROBERT P, LEHERICY S, GRAND S, et al. T1-weighted hypersignal in the deep cerebellar nuclei after repeated administrations of gadolinium-based contrast agents in healthy rats: difference between linear and macrocyclic agents[J]. Invest Radiol,2015,50(8):473-480. doi: 10.1097/RLI.0000000000000181 [5] ROBERT P, VIOLAS X, GRAND S, et al. Linear gadolinium-based contrast agents are associated with brain gadolinium retention in healthy rats[J]. Invest Radiol,2016,51(2):73-82. doi: 10.1097/RLI.0000000000000241 [6] LOHRKE J, FRISK A L, FRENZEL T, et al. Histology and gadolinium distribution in the rodent brain after the administration of cumulative high doses of linear and macrocyclic gadolinium-based contrast agents[J]. Invest Radiol,2017,52(6):324-333. doi: 10.1097/RLI.0000000000000344 [7] FDA. Gadolinium-based contrast agents for magnetic resonance imaging (MRI): drug safety communication—FDA evaluating the risk of brain deposits with repeated use[EB/OL]. (2017-05-23)[2019-08-13] https://www.fda.gov/safety/medwatch/safetyinformation/safetyalertsforhumanmedicalproducts/ucm456012.htm. [8] E M Agency.PRAC concludes assessment of gadolinium agents used in body scans and recommends regulatory actions, including suspension for some marketing authorisations. pdf[EB/OL]. (2017-03-10)[ 2019-08-13] http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2017/03/WC500223209. [9] 食药监总局. 总局提示关注含钆对比剂反复使用引起脑部钆沉积的风险[EB/OL].(2017-12-06)[2019-08-13].http://samr.cfda.gov.cn/WS01/CL0050/218164.html [10] 食药监总局. 药品不良反应信息通报(第76期) 关注含钆对比剂反复使用引起脑部钆沉积风险[EB/OL]. (2017-12-06)[2019-08-13]. http://samr.cfda.gov.cn/WS01/CL1989/218163.html [11] FRENZEL T, LENGSFELD P, SCHIRMER H, et al. Stability of gadolinium-based magnetic resonance imaging contrast agents in human serum at 37 degrees C[J]. Invest Radiol,2008,43(12):817-828. doi: 10.1097/RLI.0b013e3181852171 -

 点击查看大图
点击查看大图
图(1) / 表(5)
计量
- 文章访问数: 4117
- HTML全文浏览量: 1889
- PDF下载量: 33
- 被引次数: 0
