

 下载:
下载:
 下载:
下载:

-
根据WHO的数据,每年约有399 000人死于与丙型肝炎病毒(hepatitis C virus, HCV)感染有关的肝病。目前,尚没有疫苗可用于预防HCV感染。HCV是黄病毒科中一种有包膜的正链单链RNA病毒。HCV基因组RNA编码为一种多聚蛋白,这种多聚蛋白可以被宿主和病毒的蛋白酶切割转化为成熟蛋白,其中包括:核心蛋白、糖蛋白E1和E2(组成结构蛋白)、离子通道p7和非结构蛋白,包括NS2、NS3、NS4A、NS4B、NS5A和NS5B等。非结构病毒蛋白NS3/4A、NS4B、NS5A和NS5B组装并与宿主蛋白相互作用,形成病毒复制复合物。非结构蛋白之一的NS5B是一种HCV复制酶,它也是一种RNA依赖的RNA聚合酶(RdRp),可以复制病毒RNA的基因组[1]。因此,NS已经成为药物开发的主要目标[2]。这类以NS蛋白为靶点的抗丙肝药物,可以直接杀死HCV,也称为直接抗病毒药,例如,蛋白酶抑制剂特拉匹韦(telaprevir)或西咪匹韦(simeprevir)和RNA聚合酶抑制剂索磷布韦(sofosbuvir)对大多数常见HCV基因型的有效率高达90%,从而提高了持续的病毒学应答率。但是,这些直接抗病毒药治疗可能会导致高昂的治疗费用以及一些其他问题:如耐药性、药物相互作用,以及疲劳、头痛、恶心、失眠、甚至贫血等不良反应。因此,针对HCV生命周期不同阶段的新型药物可能为抗HCV耐药性发展和感染复发等提供有前途的方法。
从天然产物中寻找和发现新的活性化合物一直是新药研究的主要方向之一。近些年来,具有抗HCV作用的天然化合物越来越受到重视。本文对近年来报道的具有较好活性的天然抗HCV活性化合物做一综述,为天然产物抗HCV研究提供一些思路。
-
NS3作为HCV的一种重要的非结构蛋白,在HCV复制过程中起着重要的作用。NS3是一种丝氨酸蛋白水解酶,也是病毒解旋酶,可以解螺旋RNA×RNA和RNA×DNA二倍体,对病毒复制是非常重要的酶。因此,以NS3为靶标寻找抗HCV化合物也是抗HCV药物研究重要方向之一(图1)。Lee等[3]报道从中药桑白皮的乙醇提取物中分离鉴定了一类2-芳基苯并呋喃类化合物。其中moracin P(1)和moracin O(2)在这类化合物中显示出对HCV较强的抑制活性,IC50分别为35.6和80.8 μmol/L;对NS3的抑制活性IC50分别为42.9和27.0 μmol/L。
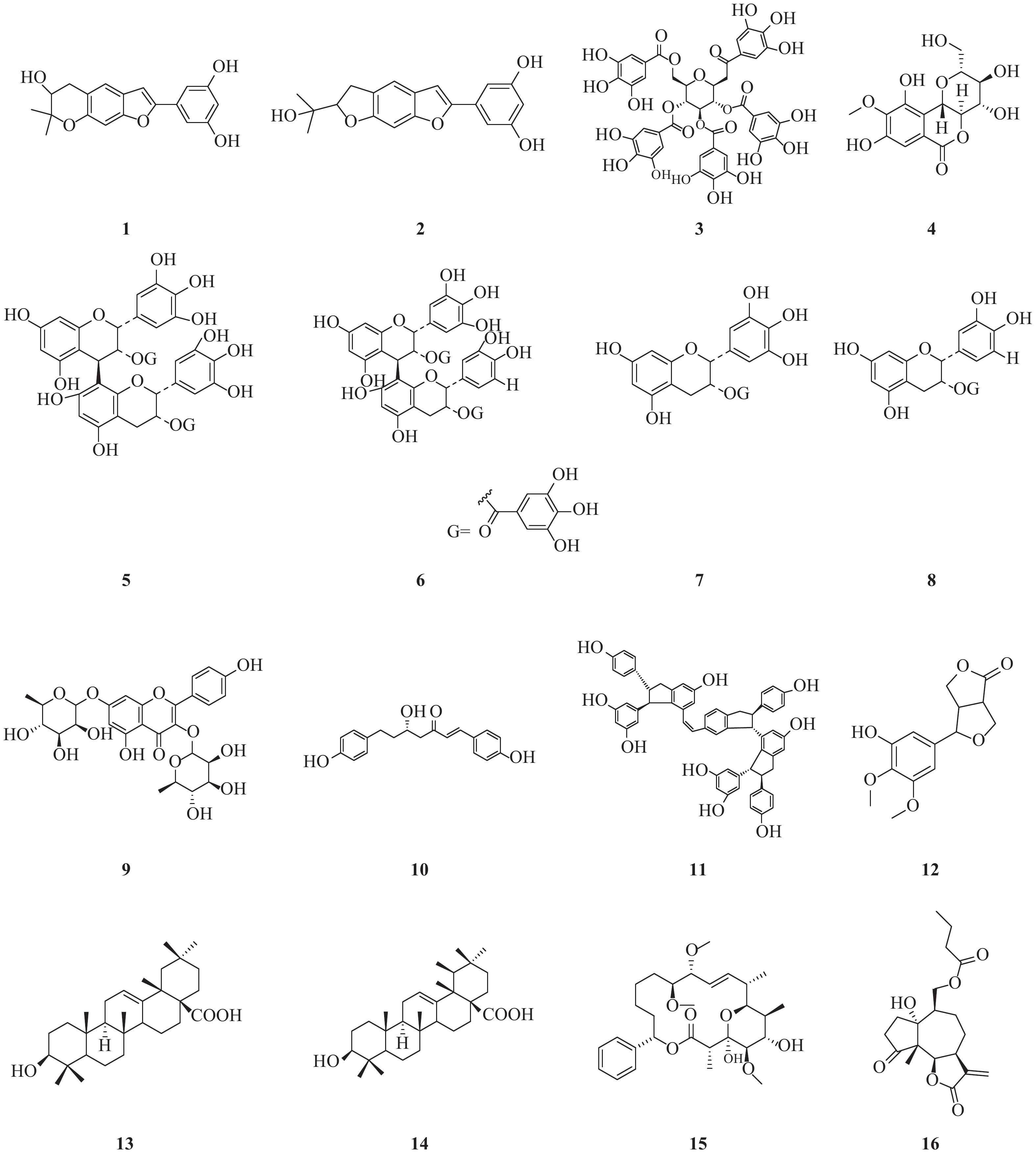
图 1 干扰HCV复制的天然产物化学结构
Zuo等报道了从黑蕊虎耳草(Saxifraga melanocentra Franch)中分离得到了一类具有HCV的NS3丝氨酸蛋白水解酶抑制活性的多酚类化合物[4]。其中化合物1,2,3,4,6-五-O-没食子酰-β-D-葡萄糖苷(3)、1,4-二-O-没食子酰-3,6-(R)-六羟基二苯甲酰基-β-D-吡喃葡萄糖(石榴皮鞣质)、1,2,4,6-四-O-没食子酰-β-D-葡萄糖苷、1,2,4-三-O-没食子酰-3,6-六羟基二苯甲酰基葡萄糖(石榴叶鞣质)和1,3,6-四-O-没食子酰-β-D-葡萄糖苷,对NS3丝氨酸蛋白水解酶抑制活性IC50分别为0.68、0.76、0.81、0.85和1.01 μmol/L。没食子酸、没食子酸甲酯、虎耳草素(4)、山奈酚、山奈酚-3-O-β-D-葡萄糖苷和2′-O-没食子酰芦丁,对NS3丝氨酸蛋白水解酶抑制作用IC50分别为1.76、1.36、1.71、1.61、1.03和4.86 μmol/L。而槲皮素和芦丁对NS3的作用相对较弱,IC50为33.11和77.05 μmol/L。从构效关系可以发现,较多的没食子酰结构有利于提高抑制NS3活性。
Zuo等[5]报道了从狭叶红景天Rhodiola kirilowii (Regel) Maxim中分离得到了4个(-)-表儿茶素衍生物:红景天素(5)、3,3′-二没食子酰基原花青素(6)、(-)-表没食子儿茶素-3-O-没食子酸酯(7, EGCG)和(-)-表儿茶素-3-O-没食子酸酯 (8, ECG),它们均显示出较好的抗HCV NS3丝氨酸蛋白水解酶活性,其IC50分别为0.77、0.91、8.51和18.55 μmol/L。从这两个研究结果可以发现,没食子酚或者其他具有较多酚羟基结构的天然多酚类化合物具有较好的NS3丝氨酸蛋白水解酶抑制活性。
从中药桑寄生醇提物的乙酸乙酯部位分离出10种化合物,其中山奈酚-3,7-双鼠李糖苷(19.4 μmol/L, 9)和(3S)-3-羟基-1,7-双(4-羟基-苯基)-6E-庚烯-5-酮(28.7 μmol/L, 10)显示出最强的抗HCV NS3蛋白酶抑制活性,其他分离得到的黄酮类化合物和二芳基庚类化合物也均具有抗HCV NS3蛋白酶抑制活性[6]。
从葡萄根提取物中得到的一种白藜芦醇四聚体vitisin B(11)显示出对HCV复制的抑制作用(IC50=6 nmol/L),而且细胞毒性较低(EC50>10 μmol/L)。与NS5B聚合酶抑制剂索磷布韦(sofosbuvir)联合使用时,vitisin B显示出协同作用。对vitisin B耐药的HCV变异株研究表明,NS3解旋酶为其潜在靶标。vitisin B可以和纯化的NS3解旋酶在体外直接结合,而显示出强效HCV NS3解旋酶抑制作用(IC50 = 3 nmol/L)。进一步的体内药动学实验研究表明,腹腔注射vitisin B后肝脏可以达到临床上可接受的浓度[7]。
Wu等[8]报道了从楝科植物大叶桃花心木(Swietenia macrophylla)枝干中分离得到化合物3-hydroxy caruilignan C (3-HCL-C, 12) 在蛋白和RNA水平均显示出较强的抗HCV活性,EC50 = 10.5 μmol/L。3-HCL-C与IFN-α、NS5B聚合酶抑制剂NM-107(2′-C-methylcytidine),或者NS3/4A蛋白水解酶抑制剂特拉匹韦联合使用,可以显著增强对HCV的RNA复制的抑制作用。
文献报道[9]五环三萜中的齐墩果酸(13)和熊果酸(14)能显著抑制HCV复制,IC50分别为2.9、10.6 μg/ml。而且这种作用部分是通过非竞争性抑制NS5B依赖性RNA聚合酶而产生。
从粘杆菌的次生代谢产物中得到的化合物soraphen A(15),具有显著的抗HCV活性,EC50为2.30 nmol/L。soraphen A既不抑制HCV RNA翻译,也不抑制HCV入侵,而是抑制HCV复制,可能是通过抑制HCV复制位点的膜质网发挥作用[10]。
Hu等从美洲小白菊(Parthenium hispitum Raf)中分离鉴定了9个C14-氧代1α-羟基-11(13)-伪愈创木烯6β,12-内酯类化合物,显示出较好的抑制HCV复制的作用[11]。其中化合物16在2 μmol/L浓度对HCV的抑制率可达90%。
-
HCV入侵正常肝细胞,是HCV完成感染正常肝细胞的最初阶段。如果可以阻止病毒对肝细胞的入侵,那么就可以将HCV“拒之门外”,从而起到保护正常细胞的作用。由于大多数药物开发策略都针对病毒生命周期的复制阶段,因此对于研究HCV入侵抑制剂是抗HCV药物的研究重要方向之一(图2),尤其是在接受肝移植的患者中。
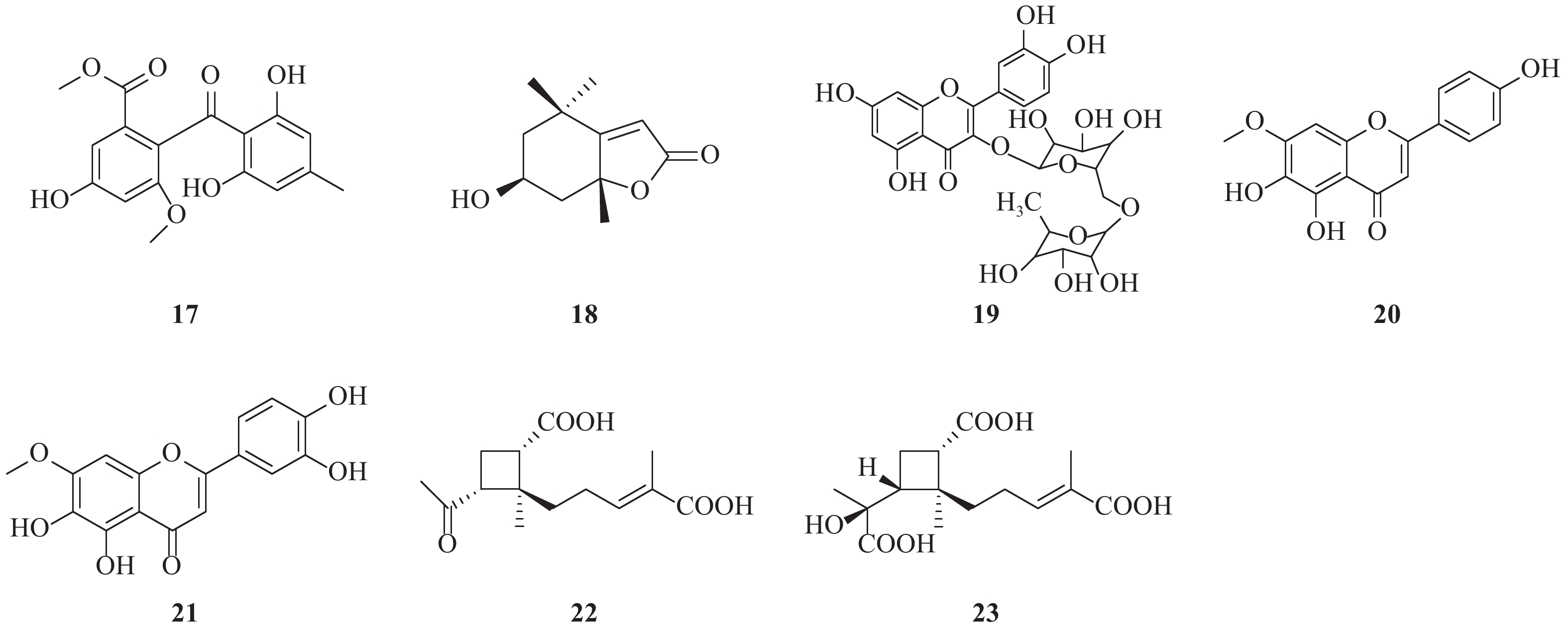
图 2 抑制病毒入侵的天然产物化学结构
Nakajima等[12]从海藻、苔藓等分离出的真菌菌株的代谢产物中,发现了sulochrin(17)具有较好的抑制HCV活性。sulochrin是一种funicone类衍生物,可以以剂量依赖关系降低HCV对细胞的感染率和HCV核心蛋白的表达,与空白对照相比,可以降低至其1/3~1/4水平。而且,在浓度达50 μmol/L时,仍没有表现出明显的细胞毒性。进一步采用HCVpp进行实验,结果表明sulochrin可以抑制病毒入侵正常细胞。sulochrin与干扰素或者特拉匹韦联合使用,可以显著增强对抗HCV作用。构效关系研究表明,1,3-二羟基-5-甲基苯基结构是重要的药效基团。
从植物叶下珠(Phyllanthus urinaria)分离得到的一种单萜类内酯地芸普内酯(LOD, 18)是一种新型HCV抑制剂。LOD可以有效抑制有利病毒颗粒,抑制病毒黏附和阻止病毒入侵和融合,而对病毒复制或翻译影响以及I型IFN宿主抗病毒免疫反应影响很小。基于酶联免疫吸附测定(ELISA)的结合分析,证实了单萜类化合物能够有效阻断HCV颗粒黏附于宿主细胞表面的能力。此外,LOD可以抑制几种HCV基因型菌株的感染。LOD可能成为抗HCV药物的潜在候选药物[13]。
Qian等[14]研究发现从五味子中分离得到的五环三萜酸甘五酸(SZA)能有效抑制HCV入侵肝细胞,却不会干扰病毒在细胞表面的结合或内在化。但是,在SZA的作用下,细胞融合过程会受到损害,并且宿主膜的流动性增加。此外,还发现SZA可抑制HCV向邻近细胞的扩散,并且SZA与干扰素或特拉普韦的联合使用对抗HCV活性具有协同效应。除此之外,SZA能够抑制体内HCV感染。SZA靶标不同于常规的抗病毒药物。因此,SZA是一种用于HCV抑制剂的潜在治疗性化合物,尤其是对于在肝移植过程中需要预防HCV再感染的患者。
Bose等[15]以保肝作用闻名的水果为研究对象,从中筛选寻找HCV入侵抑制剂,发现蔷薇科李属植物欧洲李中分离得到的黄酮类化合物芦丁具有抗HCV作用。研究表明,芦丁(19)能够抑制HCV-LP(丙型肝炎病毒样颗粒)与肝癌细胞的结合,并抑制细胞培养来源的HCV(HCVcc)进入肝癌细胞。重要的是,芦丁对肝癌细胞没有毒性。此外,芦丁可以直接作用于病毒颗粒来抑制HCV生命周期的早期侵入阶段。因此,芦丁是一种用于治疗HCV感染的候选药物。
从南美植物Pterogyne nitens中分离得到了两种具有抗HCV作用的类黄酮化合物:对HCV复制周期有影响的珍珠梅素(sorbifolin, 20)和胡麻黄素(pedalitin, 21)。化合物20和21能够在非细胞毒性条件下抑制病毒进入高达45.0%和78.7%。胡麻黄素通过对病毒颗粒的直接作用和干扰,阻止病毒进入宿主细胞。同样,珍珠梅素也有抗HCV作用[16]。
从海绵真菌哈茨木霉菌(Trichoderma harzianum)中分离出两个新型倍半萜烯的类似物:哈兹烷酸A(harzianoic acids A, 22)和B(harzianoic acids B, 23)。 两种化合物均显示出降低HCV RNA水平的抑制活性,且细胞毒性较低。哈兹烷酸A的EC50=24.5 μmol/L,CC50=75.3 μmol/L;哈兹烷酸B的EC50=20.4 μmol/L,CC50=74.3 μmol/L。作用机制表明,这些化合物可以阻断HCV生命周期的侵入,而病毒E1/E2和宿主细胞CD81是潜在的靶蛋白[17]。
-
近年来,除了报道了一些已经初步明确了作用阶段或机制的天然产物外,还报道了一些未明确阶段和机制的化合物(图3)。Li等报道了从新疆紫草(Arnebia euchroma)分离得到的紫草素(24)显示出较好的抗HCV作用,其EC50= 0.025 μg/ml,CC50= 1.089 μg/ml,选择指数(SI)= 43.56[18]。
图 3 具有抗HCV作用的天然产物化学结构
Adianti通过活性导向分离法从甘草中提取出的甘草香豆素(25)、甘草宁(26)、甘草酚(27)、甘草素(28)均具有较好抗HCV活性,IC50分别为8.8、7.2、4.6和16.4 mg/ml。但是,甘草的主要化学成分甘草酸以及甘草酸铵仅具有很弱的抗HCV活性。此外,甘草查耳酮A(licochalcone A)和光甘草定(glabridin)也显示出较好抗HCV活性,IC50分别为2.5和6.2 mg/ml。另一个查耳酮异甘草素也显示出抗HCV活性,IC50为3.7 mg/ml[19]。
由黏杆菌Labilithrix luteola(DSM 27648 T)分离而来的两种新型次生代谢产物,labindole A [2-甲基-3-(2-硝基乙基)-3H-吲哚, 29]和labindole B [2-甲基-3-(2-硝基乙烯基)-3H-吲哚, 30],以及已知的5种代谢物经生物活性检测发现:-氯-9H-咔唑(34)具有显著的抗HCV活性。9H-咔唑(33)和labindoles A(31)和B(32)都具有一定的HCV抑制作用,其中4-羟甲基喹啉(35)活性较弱,并且均没有细胞毒性[20]。
两种新型的乌苏烷型三萜皂苷bodiniosides O(36)和bodiniosides P(37)在体外均表现出一定的抗HCV活性,选择性指数分别为30.63和9.08。在所测试的化合物中,化合物36表现出一定抗HCV活性,EC50值为0.41 nmol/L,CC50值为12.56 nmol/L,SI值为30.63。此外,化合物37也具有较好的抗HCV活性,EC50值为1.58 nmol/L,CC50值为14.35 nmol/L,SI值为9.08[21]。
从豆科槐属植物苦豆子(Sophora alopecuroides)中提取的aloperine(38)是一种具有独特的内环结构的喹喔啉生物碱。研究发现其具有一定的抗HCV活性,最大有效浓度(EC50)为4.32 μmol/L,选择指数(SI)为54.5[22]。
从sarcocornia甲醇水提物中分离出一种新的黄酮醇三糖苷,鼠李糖3-O-2G-鼠李糖苷或鼠李糖苷3-O-(2″,6″-O-α-二鼠李糖基)-β-葡萄糖苷(39)。鼠李糖苷三糖苷被证实具有显著的抗HCV活性,IC50值为8.9 μmol/L[23]。
-
综上所述,近年来从天然产物中发现了大量的抗HCV活性结构,结构类型主要包括黄酮、萜类、木质素等。这些活性天然产物可以干扰HCV复制,抑制病毒入侵等,并且毒性较低。因此,从天然产物中寻找和发现的抗HCV活性结构对于抗HCV药物的研究具有重要意义。
Research progress of natural products against hepatitis C virus
-
摘要: 丙型肝炎病毒(HCV)是导致慢性肝炎、肝硬化和肝细胞癌的重要因素。当前,没有疫苗可用于预防HCV感染。该文综述了近年来从植物中发现的具有抗HCV活性的天然产物,并依据其作用机制进行了分类阐述和总结,以期为从天然产物中发现抗HCV药物提供依据。Abstract: Hepatitis C virus (HCV) is an important factor leading to chronic hepatitis, cirrhosis and hepatocellular carcinoma. Currently, there is no vaccine available to prevent HCV infection. This article reviews the natural products with anti-HCV activity discovered from plants in recent years, and classifies and summarizes them according to their mechanism of action, in order to provide a basis for discovering anti-HCV drugs from natural products.
-
Key words:
- hepatitis C virus /
- natural products /
- virus replication /
- virus entry /
- research progress
-
在正常条件下,大部分肝细胞处于静止状态(G0期),通常很少进入有丝分裂期。一旦肝细胞受到手术切除和药物等应激的刺激,它们会迅速对促分裂原产生反应,过渡到有丝分裂期,并开始增殖[1-4]。肝细胞增殖不仅是肝脏自我修复和再生的基础,也是应对慢性肝炎、肝硬化以及药物性肝损伤(DILI)等多种损害的重要机制。研究表明,肝脏出现病理性损伤后产生多种肝细胞因子,例如纤维蛋白原样蛋白(FGL1)、Hepassocin、肝细胞生长因子(HGF)等,在促进肝细胞增殖方面发挥重要作用[5-8]。例如,FGL1通过激活细胞增殖相关的信号通路促进肝细胞的再生,从而加速肝脏的修复[9]。此外,Hepassocin通过自分泌机制促进肝细胞生长[6]。这些研究提供了肝脏疾病治疗的新思路,尤其是调控这些肝细胞因子来加速肝功能的恢复。
Orosomucoid-1(ORM1),也称为α-1-酸性糖蛋白1(AGP1),作为一个已知的肝细胞因子[10],是一种主要由肝脏合成分泌的急性期蛋白,在应激条件下(如组织损伤和炎症)显著升高,具有调控细胞外基质(ECM)、调节糖代谢、维持血管通透性等重要的生物学功能[10-12]。此外,ORM1在结肠腺癌细胞、肝细胞、免疫细胞等多种细胞上,表现出促进增殖的作用[13-15]。但ORM1调控肝细胞增殖的具体作用机制尚不完全明确,有待进一步研究。
为进一步探究ORM1在肝细胞增殖中的具体作用及机制,本研究首先在两种肝细胞系上验证了ORM1促进肝细胞增殖,并通过对ORM1敲除小鼠的肝脏组织测序,挖掘ORM1调控肝细胞增殖的可能机制。本研究旨在揭示ORM1促进肝细胞增殖的潜在功能和机制,为肝脏损伤修复及相关疾病的治疗提供新的分子靶点和理论依据。
1. 材料与方法
1.1 试剂
α1-酸性糖蛋白(美国 Sigma-Aldrich 公司)、CCK8试剂盒(Adamas life)、Lipofectamine 3000试剂(Thermo Fisher Scientific)。
1.2 实验细胞
小鼠肝癌细胞系Hepa1-6,购自中国科学院细胞库。培养液配方:DMEM、10%FBS、1%双抗生素。培养条件:5%CO2、37 ℃恒温孵育箱。人肝癌细胞系HepG2,购自武汉普诺赛生命科技有限公司。培养液配方:MEM(含NEAA)、10%FBS、1%双抗生素。培养条件:5%CO2、37 ℃恒温孵育箱。
1.3 CCK8
细胞接种于96孔板,密度约104/孔,每组复孔为9;ORM1过表达、干扰或人源ORM给药(10、50、100 μg/ml)处理相应时间后,弃去培养基,在细胞贴壁后,使用无血清培养基处理细胞,培养相应时间后,加入CCK8试剂,吸光度测定(Epoch BioTeK酶标检测仪)450 nm处的A值,根据不同组之间的A值计算组间差异。
1.4 细胞瞬时转染
通过LipofectamineTM
3000 将ORM1过表达质粒(序列见NM_008768.2)或siRNA转染进细胞内,以实现过表达或干扰目标基因的目的。当细胞密度达到50%~70%时,进行过表达质粒的瞬时转染实验;当细胞密度达到30%~50%时,进行siRNA的瞬时转染实验。吸取舍弃原培养基后用1×PBS轻柔洗涤细胞1~2次,根据转染条件配制好转染试剂,随后将配置好含有过表达质粒(或siRNA)的DMEM培养基加入至细胞培养基中用于细胞继续培养,最后置于细胞培养箱中孵育24 h(若为转染siRNA,则孵育48 h)即可完成细胞瞬时转染试验。siRNA序列及转染试剂配制方法见表1、表2。表 1 ORM1 siRNA序列siRNA名称 siRNA序列(5'→3') si-ORM1 CCACCAACUUGAUAAACGATT 表 2 96孔培养板转染条件培养皿 Opti-MEM (μl) Lipo3000 (μl) DNA转染 RNA转染 DNA (ng) P3000 (μl) RNA (pmol) 96孔板 2×10 0.3 100 0.2 20 1.5 RNA-seq及差异基因富集分析
使用ORM1基因敲除(KO)小鼠和对应的野生型(WT)小鼠(均为C57BL/6背景,n=6)。在小鼠24周龄时采集肝脏组织,液氮速冻并存储于−80°C。RNA-seq委托上海欧易生物公司进行,差异基因分析通过DESeq完成,筛选标准为FoldChange>1.5或<0.66,且P<0.05。
差异基因富集分析通过DAVID进行,进行Kyoto Encyclopedia of Genes and Genomes (KEGG)富集分析。KEGG分析用于揭示差异基因在代谢通路中的富集情况。富集分析筛选标准为P<0.05。
1.6 数据统计与分析
实验数据采用GraphPad Prism 9.0软件进行统计与分析。单因素两组之间相互比较,采用双尾非配对t检验(Student’t)检验分析;单因素多组之间相互比较,采用单向方差分析(One-way ANOVA)检验,组间均值两两比较采用Dunnett's multiple comparisons test。实验结果均采用“均数±标准误(Mean±SE)”表示。以P<0.05认为差异具有统计学意义。
2. 结果
2.1 ORM1在小鼠肝切除术后表达增加
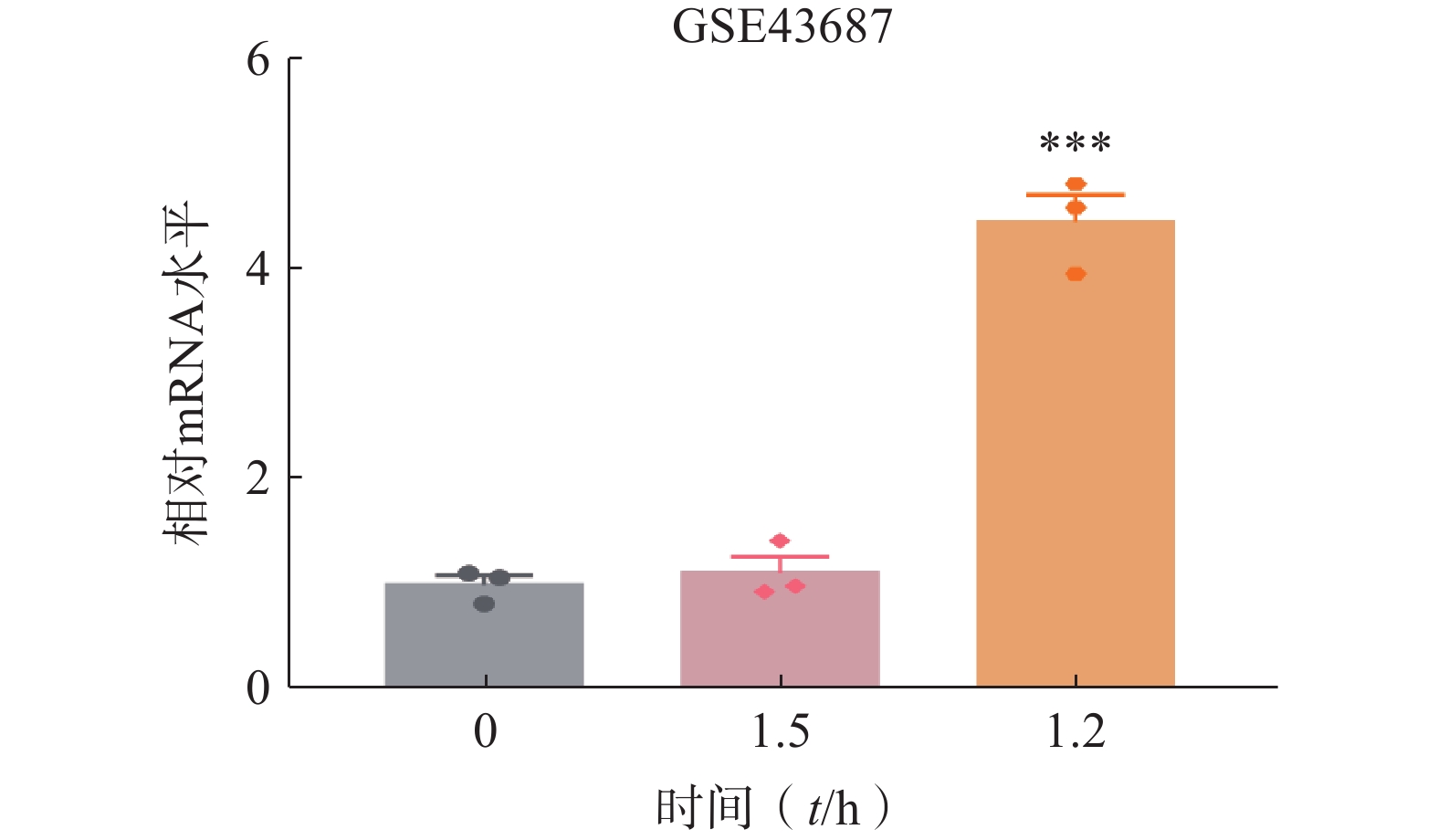
肝脏组织在肝切除术后会进入肝细胞快速增殖状态[16]。通过检索GEO数据库,选取C57BL/6小鼠在2/3肝脏部分切除术(PH)后数据(GSE43687)进行分析。结果显示,肝脏组织中ORM1的表达在PH后1.5 h时无显著变化,在PH后12 h显著上升(图1)。这一结果提示ORM1可能参与肝脏再生过程。
2.2 ORM1促进小鼠和人肝细胞增殖
为验证ORM1对肝细胞增殖的影响,我们在小鼠和人的肝细胞系上进行了ORM1过表达、干扰或ORM外源性给药实验。在小鼠Hepa1-6肝癌细胞系过表达ORM1(pCMV-ORM1),与对照质粒组(pCMV)组相比,转染后第3、4天ORM1过表达质粒组的细胞增殖显著增加(图2A)。而干扰ORM1后细胞增殖能力呈现相反的结果(图2B)。人HepG2肝细胞上给予不同浓度外源性ORM,48 h后检测细胞增殖情况,可见随着ORM1浓度的增加,细胞增殖呈现显著上升(图2C)。上述结果表明ORM1对肝细胞增殖具有显著的促进作用。
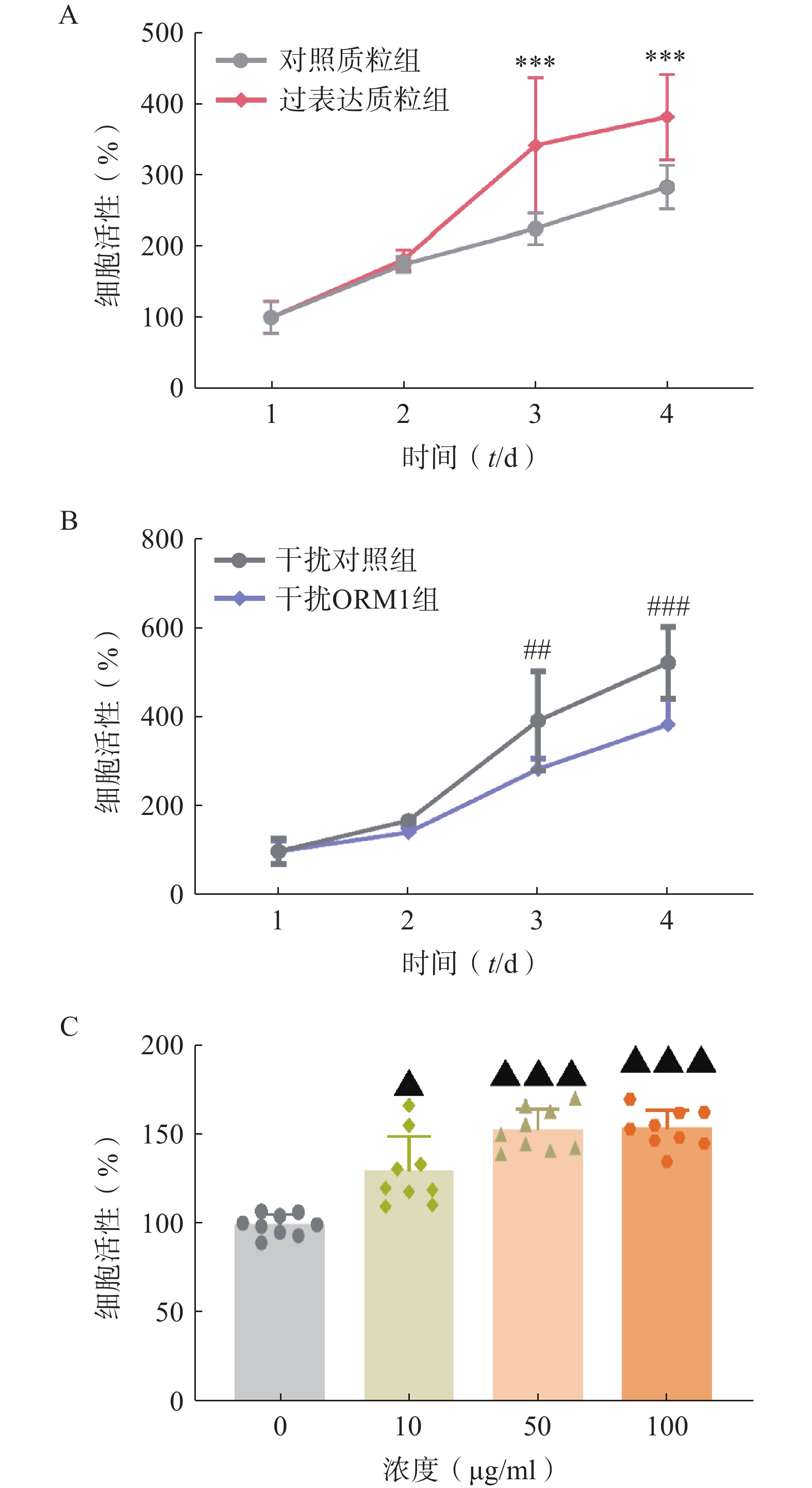 图 2 ORM1过表达、干扰或外源性给药对细胞增殖的影响A. ORM1过表达对肝细胞增殖的影响,过表达ORM1质粒组与对照质粒组在转染后第1天至第4天测定细胞活力百分比(n=9);B. ORM1干扰对肝细胞增殖的影响,干扰ORM1组与干扰对照组在转染后第1天至第4天测定细胞活力百分比(n=9);C. 外源性ORM给药对肝细胞增殖的影响,不同浓度ORM处理48h后检测细胞增殖情况,细胞增殖以百分比形式表示(n=9)***P<0.001,与对照质粒组比较;##P<0.01,###P<0.001,与干扰对照组比较;▲P<0.05,▲▲▲P<0.001,与0 μg/ml组比较。
图 2 ORM1过表达、干扰或外源性给药对细胞增殖的影响A. ORM1过表达对肝细胞增殖的影响,过表达ORM1质粒组与对照质粒组在转染后第1天至第4天测定细胞活力百分比(n=9);B. ORM1干扰对肝细胞增殖的影响,干扰ORM1组与干扰对照组在转染后第1天至第4天测定细胞活力百分比(n=9);C. 外源性ORM给药对肝细胞增殖的影响,不同浓度ORM处理48h后检测细胞增殖情况,细胞增殖以百分比形式表示(n=9)***P<0.001,与对照质粒组比较;##P<0.01,###P<0.001,与干扰对照组比较;▲P<0.05,▲▲▲P<0.001,与0 μg/ml组比较。2.3 ORM1基因敲除对肝脏细胞生物学功能及信号通路的影响
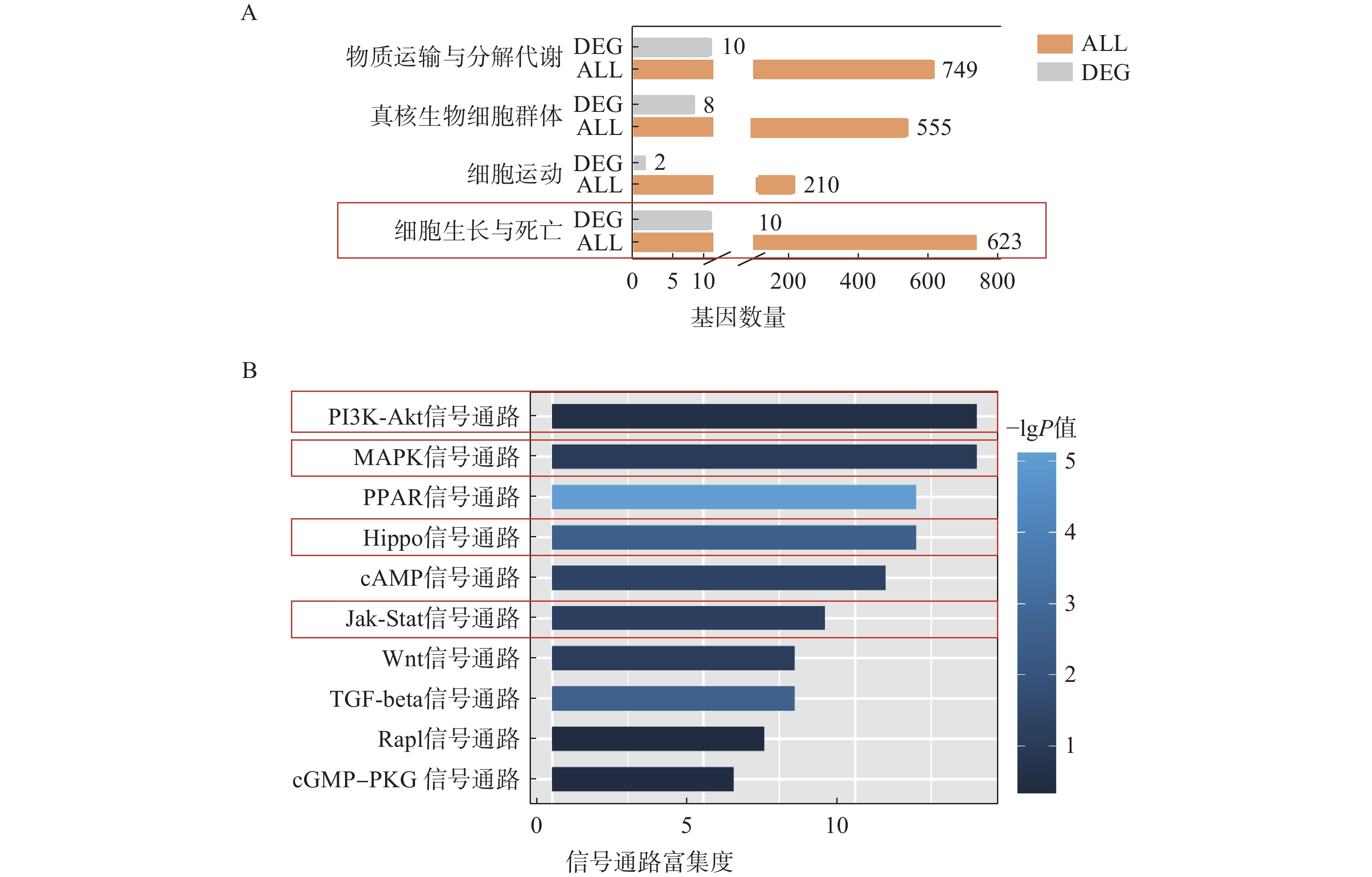
为了进一步探究ORM1基因敲除后对肝脏组织的影响,我们对ORM1基因敲除(KO)小鼠和野生型(WT)小鼠的肝脏组织进行了测序。KEGG富集分析表明,在ORM1基因敲除小鼠与野生型小鼠的比较中,差异表达基因被富集在“细胞生长与死亡”和“细胞运输与自噬”等通路中(图3A),提示ORM1缺失会引起细胞生长和死亡相关的通路相关改变。
进一步信号通路的富集分析显示,PI3K-Akt、MAPK、PPAR、Hippo、Jak-Stat等通路在差异基因表达分析中有显著富集(图3B),其中,PI3K-Akt、MAPK、Hippo、Jak-Stat通路据报道与细胞增殖高度相关[17-19]。这些结果提示,ORM1基因敲除可能影响细胞增殖关键通路,通过调控细胞生长、死亡相关的基因影响肝细胞增殖。
2.4 ORM1可能通过调控多条信号通路促进肝细胞增殖
为进一步明确ORM1在这些经典通路中的调控机制,我们重点分析了其中与细胞增殖相关的差异基因表达变化。在PI3K-Akt信号通路中,具有促增殖作用的Itgb8和Ntf3显著下调(图4A)。在MAPK信号通路中,同样具有促增殖作用的Map3k6在KO组中显著下调,而抑制细胞增殖作用的Gadd45a则显著上调,Hspa1b和Hspa2下调(图4B)。与之类似,Jak-Stat信号通路中的Il6ra和Lepr具有促增殖作用,其表达在KO组中也显著下调,而有抑制细胞增殖作用的Cish在KO组中的表达显著上调(图4C)。在Hippo信号通路中,具有促增殖作用的Tef7、Tead1、Ctgf在KO组中的表达显著下调(图4D)。这些结果表明,ORM1可能调控PI3K-Akt、MAPK、Jak-Stat和Hippo信号通路中的影响细胞增殖的多种关键信号分子影响肝细胞的增殖。
 图 4 ORM1对细胞增殖相关信号通路的影响A. ORM1调控PI3K-Akt信号传导通路;B. ORM1调控MAPK信号传导通路;C. ORM1调控Jak-Stat信号传导通路;D. ORM1调控Hippo信号传导通路(n=6)*P<0.05,**P<0.01,***P<0.001,与ORM1+/+组比较。
图 4 ORM1对细胞增殖相关信号通路的影响A. ORM1调控PI3K-Akt信号传导通路;B. ORM1调控MAPK信号传导通路;C. ORM1调控Jak-Stat信号传导通路;D. ORM1调控Hippo信号传导通路(n=6)*P<0.05,**P<0.01,***P<0.001,与ORM1+/+组比较。3. 讨论
本研究中,我们揭示了ORM1在肝细胞增殖中的潜在调控机制。首先,利用GEO数据库(GSE43687)分析,发现ORM1在小鼠2/3肝切除术后12h表达升高,提示ORM1可能参与肝脏再生。而在体外肝细胞系中,过表达ORM1或外源性给药ORM,可以促进肝细胞增殖;相反,干扰ORM1抑制的肝细胞的增殖,进一步验证了ORM1对肝细胞增殖的促进作用。此外,我们对ORM1基因敲除小鼠组学分析,发现PI3K-Akt、MAPK、Hippo、Jak-Stat等细胞增殖相关通路的多个信号分子在ORM1缺失时表现出显著变化,提示这些关键通路可能参与了ORM1对肝细胞增殖的调控。
在PI3K-AKT信号通路中,Ntf3(neurotrophin-3)主要通过与受体TrkC结合,促进细胞的存活、增殖和分化[20]。ORM1缺失后Ntf3的下调可能导致细胞存活和再生能力减弱。而Itgb8(整合素β8)和Lamc2(laminin subunit gamma-2)作用类似,均是通过与细胞外基质(ECM)蛋白(如纤连蛋白、胶原蛋白等)结合,介导细胞与其周围环境的相互作用,从而影响细胞的增殖和分化[21-22]。ORM1缺失后,Itgb8和Lamc2下调提示ECM重塑减弱,从而无法为新生肝细胞的重新附着与生长提供必要的微环境支持。
在MAPK信号通路中,Gadd45a(growth arrest and DNA damage-inducible alpha)是一种生长停滞和DNA损伤诱导因子,通过抑制细胞周期相关激酶(如CDK1和CDK2),阻止细胞从G1期或G2/M期进入下个阶段[23]。ORM1缺失后Gadd45a上调提示可能出现细胞周期停滞,从而抑制了肝细胞的增殖。Map3k6(mitogen-activated protein kinase 6)作为一种激酶,通过磷酸化MAPK激酶调控下游的信号分子和后续的细胞生物学反应,如细胞增殖、分化、存活、迁移和应激反应[24]。ORM1缺失后Map3k6下调可能导致MAPK通路激活减少。Hspa1b和Hspa2是热休克蛋白,在抑制凋亡通路主要通过抑制线粒体凋亡途径,caspase的活化以及调控JNK信号通路发挥作用;同时,Hspa1b和Hspa2通过清除活性氧(ROS)和稳定线粒体功能,发挥抗氧化应激损伤的作用,进而增加细胞存活。Hspa1b和Hspa2的下调提示ORM1缺失可能削弱细胞的抗氧化应激能力,从而影响肝细胞存活[26]。
此外,Jak-Stat3信号通路激活通过诱导与细胞增殖和抗凋亡相关的基因表达,如Cyclin D1和Bcl-2,从而促进细胞增殖、存活和组织修复[27]。而Cish(cytokine-inducible SH2-containing protein)是该通路一个重要的负反馈调节因子,通过抑制Jak-Stat信号的活化,调控细胞因子信号传导的强度和持续时间,发挥抑制细胞增殖和促进细胞凋亡的作用[28]。与之相反,Il6ra是IL-6受体的亚基,IL-6结合IL-6受体后激活Jak-Stat3信号通路,诱导与细胞存活、增殖和组织修复相关基因的表达[29]。此外,Lepr(leptin receptor)是瘦素(leptin)的主要受体,该受体激活后亦可激活下游Jak-Stat3信号通路[27]。特别是在肝脏中,Lepr信号能够促进肝细胞的再生和修复能力[30]。而ORM1敲除时,Cish上调,Il6ra、Lepr下调均表明JAK-STAT通路激活减少,从而导致细胞增殖减弱。
最后,在Hippo信号通路中,Tcf7在Hippo信号通路中扮演着重要的角色,其与细胞增殖的关系主要通过YAP/TAZ的激活来实现,并参与促进细胞增殖[31],Ctgf是一种与细胞外基质(ECM)相关的结合肝素的蛋白,能够直接与整合素结合。它由成纤维细胞合成,并能促进细胞的增殖和趋化[32],ORM1敲除导致Hippo通路中Tcf7和Ctgf表达下调,可能通过解除其对肝细胞增殖的抑制作用而促进细胞增殖。ORM1缺失还导致了该通路Tead1基因的下调,而Tead1作为YAP(yes-associated protein)和TAZ的下游效应器,与YAP/TAZ结合后在细胞核中调控靶基因Birc5、Cyr61、CTGF和Myc等的表达,进而促进细胞增殖、抑制细胞凋亡,并推动组织生长和再生[33]。
本研究阐释了ORM1可能通过调控PI3K-Akt、MAPK、Hippo、Jak-Stat等信号通路发挥促进肝细胞增殖的作用。提示ORM1可能通过影响肝细胞存活、ECM重塑等一系列复杂机制调节肝细胞增殖过程。未来研究应进一步探讨ORM1在这些信号通路中的具体调控机制,特别是在不同病理条件下如何影响肝细胞的增殖、凋亡以及组织修复,为肝脏疾病的治疗提供新的潜在靶点。
-
[1] BANERJEE A, RAY R B, RAY R. Oncogenic potential of hepatitis C virus proteins[J]. Viruses,2010,2(9):2108-2133. doi: 10.3390/v2092108 [2] BEAULIEU P L. Non-nucleoside inhibitors of the HCV NS5B polymerase: progress in the discovery and development of novel agents for the treatment of HCV infections[J]. Curr Opin Investig Drugs,2007,8(8):614-634. [3] LEE H Y, YUM J H, RHO Y K, et al. Inhibition of HCV replicon cell growth by 2-arylbenzofuran derivatives isolated from Mori Cortex Radicis[J]. Planta Med,2007,73(14):1481-1485. doi: 10.1055/s-2007-990249 [4] ZUO G Y, LI Z Q, CHEN L R, et al. In vitro anti-HCV activities of Saxifraga melanocentra and its related polyphenolic compounds[J]. Antivir Chem Chemother,2005,16(6):393-398. doi: 10.1177/095632020501600606 [5] ZUO G Y, LI Z Q, CHEN L R, et al. Activity of compounds from Chinese herbal medicine Rhodiola kirilowii (Regel) Maxim against HCV NS3 serine protease[J]. Antiviral Res,2007,76(1):86-92. doi: 10.1016/j.antiviral.2007.06.001 [6] YANG L Y, LIN J, ZHOU B, et al. Activity of compounds from Taxillus sutchuenensis as inhibitors of HCV NS3 serine protease[J]. Nat Prod Res,2017,31(4):487-491. doi: 10.1080/14786419.2016.1190719 [7] LEE S, YOON K D, LEE M, et al. Identification of a resveratrol tetramer as a potent inhibitor of hepatitis C virus helicase[J]. Br J Pharmacol,2016,173(1):191-211. doi: 10.1111/bph.13358 [8] WU S F, LIN C K, CHUANG Y S, et al. Anti-hepatitis C virus activity of 3-hydroxy caruilignan C from Swietenia macrophylla stems[J]. J Viral Hepat,2012,19(5):364-370. doi: 10.1111/j.1365-2893.2011.01558.x [9] KONG L B, LI S S, LIAO Q J, et al. Oleanolic acid and ursolic acid: novel hepatitis C virus antivirals that inhibit NS5B activity[J]. Antiviral Res,2013,98(1):44-53. doi: 10.1016/j.antiviral.2013.02.003 [10] KOUTSOUDAKIS G, ROMERO-BREY I, BERGER C, et al. Soraphen A: a broad-spectrum antiviral natural product with potent anti-hepatitis C virus activity[J]. J Hepatol,2015,63(4):813-821. doi: 10.1016/j.jhep.2015.06.002 [11] HU J F, PATEL R, LI B, et al. Anti-HCV bioactivity of pseudoguaianolides from Parthenium hispitum[J]. J Nat Prod,2007,70(4):604-607. doi: 10.1021/np060567e [12] NAKAJIMA S, WATASHI K, KAMISUKI S, et al. Specific inhibition of hepatitis C virus entry into host hepatocytes by fungi-derived sulochrin and its derivatives[J]. Biochem Biophys Res Commun,2013,440(4):515-520. doi: 10.1016/j.bbrc.2013.09.100 [13] CHUNG C Y, LIU C H, BURNOUF T, et al. Activity-based and fraction-guided analysis of Phyllanthus urinaria identifies loliolide as a potent inhibitor of hepatitis C virus entry[J]. Antiviral Res,2016,130:58-68. doi: 10.1016/j.antiviral.2016.03.012 [14] QIAN X J, ZHANG X L, ZHAO P, et al. A Schisandra-derived compound schizandronic acid inhibits entry of Pan-HCV genotypes into human hepatocytes[J]. Sci Rep,2016,6(1):850-858. [15] BOSE M, KAMRA M, MULLICK R, et al. Identification of a flavonoid isolated from plum (Prunus domestica) as a potent inhibitor of Hepatitis C virus entry[J]. Sci Rep,2017,7(1):39-65. doi: 10.1038/s41598-017-00075-1 [16] SHIMIZU J F, LIMA C S, PEREIRA C M, et al. Flavonoids from Pterogyne nitens inhibit hepatitis C virus entry[J]. Sci Rep,2017,7(1):359-362. doi: 10.1038/s41598-017-00506-z [17] LI B, LI L, PENG Z G, et al. Harzianoic acids A and B, new natural scaffolds with inhibitory effects against hepatitis C virus[J]. Bioorg Med Chem,2019,27(3):560-567. doi: 10.1016/j.bmc.2018.12.038 [18] LI H M, TANG Y L, ZHANG Z H, et al. Compounds from Arnebia euchroma and their related anti-HCV and antibacterial activities[J]. Planta Med,2012,78(1):39-45. doi: 10.1055/s-0031-1280266 [19] ADIANTI M, AOKI C, KOMOTO M, et al. Anti-hepatitis C virus compounds obtained from Glycyrrhiza uralensis and other Glycyrrhiza species[J]. Microbiol Immunol,2014,58(3):180-187. doi: 10.1111/1348-0421.12127 [20] MULWA L S, JANSEN R, PRADITYA D F, et al. Six heterocyclic metabolites from the myxobacterium Labilithrix luteola[J]. Molecules,2018,23(3):E542. doi: 10.3390/molecules23030542 [21] XIANG L, ZHANG L, CHEN X, et al. Ursane-type triterpenoid saponins from Elsholtzia bodinieri[J]. Nat Prod Res,2019,33(9):1349-1356. doi: 10.1080/14786419.2018.1477144 [22] ZHANG X, LV X Q, TANG S, et al. Discovery and evolution of aloperine derivatives as a new family of HCV inhibitors with novel mechanism[J]. Eur J Med Chem,2018,143:1053-1065. doi: 10.1016/j.ejmech.2017.12.002 [23] HAWAS U W, ABOU EL-KASSEM L T, SHAHER F, et al. In vitro inhibition of Hepatitis C virus protease and antioxidant by flavonoid glycosides from the Saudi costal plant Sarcocornia fruticosa[J]. Nat Prod Res,2019,33(23):3364-3371. doi: 10.1080/14786419.2018.1477153 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4808
- HTML全文浏览量: 1937
- PDF下载量: 33
- 被引次数: 0
