

 下载:
下载:


 下载:
下载:

-
他汀类药物抑制胆固醇合成途径的限速酶3-羟基-3-甲基戊二酸单酰辅酶A还原酶(HMG-CoA还原酶)的活性,抑制胆固醇的生成,上调肝细胞表面的LDL受体以加快LDL的分解代谢。阿托伐他汀以活性酸形式给药,通过被动扩散或OATP1B1转运进入肝细胞,主要由CYP3A4在肝脏和肠壁代谢阿托伐他汀,少量由CYP2C9、CYP2C19、CYP3A5和UGT1A1代谢。阿托伐他汀酸由CYP3A代谢产生邻、对位羟基活性代谢产物,在体内它们与各自的非活性内酯形式处于平衡状态。ABCB1(编码p-gp)和ABCG2(编码BCRP)介导阿托伐他汀及其代谢物从肝脏排入胆汁后消除(图1)[1]。
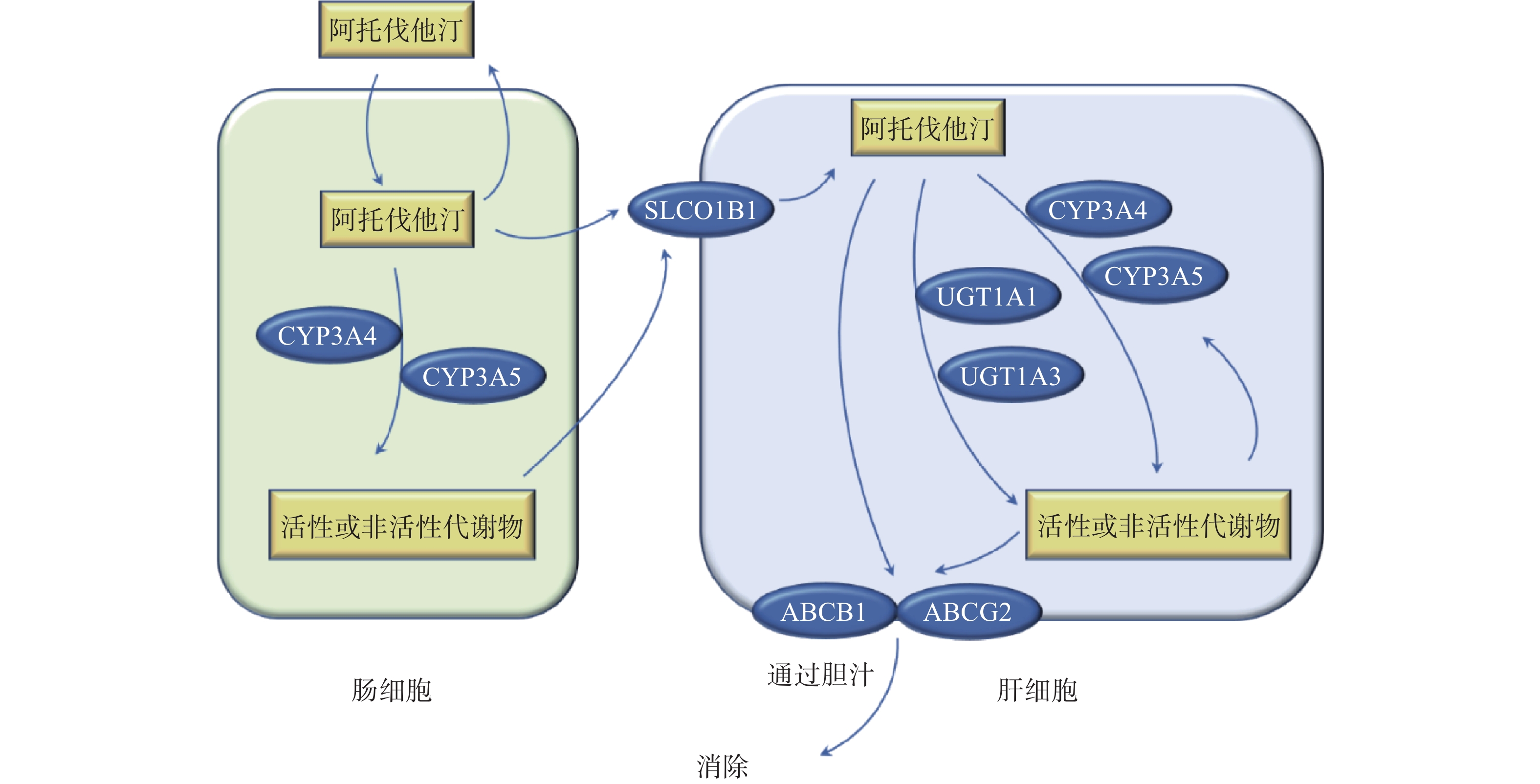
图 1 阿托伐他汀代谢途径的基因集合
-
大多数CYP3A代谢发生在肝细胞内,部分发生在小肠内。CYP3A的活性存在较大的个体间差异,显著影响阿托伐他汀的体内代谢。阿托伐他汀和阿托伐他汀内酯主要由CYP3A4同工酶代谢,CYP3A4的活性在不同个体之间可能存在10倍的差异,这可能是由于编码酶的基因多态性所致[2]。
CYP3A4*1G/*1G基因型患者的阿托伐他汀AUC0-∞比野生型和*1/*1G基因型低36%和25%。阿托伐他汀和2-羟基阿托伐他汀的tmax和(CL/Fm)在CYP3A4*1G/*1G基因型和野生型之间存在显著性差异[3]。
经阿托伐他汀治疗后,CYP3A4启动子变异(rs2740574)与野生型等位基因相比显示出更低的LDL-C水平和更好的疗效[2]。CYP3A4*1G(20230G>A)多态性对血清TC下降具有剂量依赖性(P<0.01)。携带CYP3A4*1G增加了阿托伐他汀的降脂效果,对辛伐他汀降脂治疗没有显著影响[4]。CYP3A4*22(rs35599367)会导致CYP3A4酶含量和活性显著降低,CYP3A4*22携带者的阿托伐他汀、辛伐他汀或洛伐他汀的剂量仅为野生型的20%~60%[5]。携带CYP3A4*1B(290A>G)G等位基因可降低辛伐他汀和阿托伐他汀的血药浓度,特别是女性和具有ABCB1 3435T变异等位基因的患者[6]。CYP3A4 A290G变异等位基因与阿托伐他汀治疗后较高的LDL-C水平显著相关,而非同义多态性M445T变异与治疗后较低的LDL-C水平显著相关[7]。此外,携带CYP3A4 A290G变异等位基因比野生型等位基因型的患者具有更高的HDL水平(P<0.001),可能与体内CYP3A4活性降低相关,导致阿托伐他汀活性增加[8]。临床上可减少CYP3A4*22携带者的药物剂量,CYP3A4*1B突变对他汀类药物降脂作用影响尚无定论。
-
CYP3A4和CYP3A5约占CYP450的30%,CYP3A4和CYP3A5均在肝脏和肠道中表达,其中CYP3A5主要在肝外组织中表达。CYP3A5基因与CYP3A家族其他成员一起位于7q22.1染色体上,由9个外显子组成,编码502个氨基酸。
Kivistö等[9]研究发现洛伐他汀、辛伐他汀和阿托伐他汀对CYP3A5表达者(CYP3A5*1)的疗效显著低于非表达者(CYP3A5*3)。携带CYP3A5*1等位基因的受试者1年内的平均血清TC水平和LDL-C水平分别比CYP3A5*3纯合等位基因的受试者高23%(P=0.0014)和24%(P=0.036)。在服用不依赖于CYP3A5代谢的氟伐他汀、普伐他汀的23名受试者中,未发现降血脂与CYP3A5多态性的关联。因此,CYP3A5*3突变可导致CYP3A5酶的表达降低,提高了他汀类药物的降脂疗效。
-
在192例由阿托伐他汀治疗缺血性卒中的中国患者中,Chen等[10]发现CYP2D6 rs1065852显著影响ΔLDL(P<0.001),ΔLDL/LDL (P<0.001),Δ(LDL/HDL)(P<0.001),Δ(LDL/HDL)/(LDL/HDL)(P<0.001)水平。阿托伐他汀对CYP2D6 rs1065852 G等位基因携带者具有较好的降脂效果。
-
阿托伐他汀由UGT1A1和UGT1A3介导的葡萄糖醛酸化转化为相应的内酯形式,UGT1A3是内酯化的主要酶。
Sung等[11]研究表明UGT1A3*2携带者内酯的生成量显著增加。UGT1A3*2/*2组阿托伐他汀内酯和2-羟基阿托伐他汀内酯的AUC分别比UGT1A3*1/*1组高72%和160%。UGT1A3*2携带者的TC和LDL-C降低百分比分别比非携带者低29%和18%。UGT1A3*2多态性与阿托伐他汀内酯化增加相关,可能影响其降脂作用。低表达等位基因UGT1A1*28(TA)7的携带者较正常活性等位基因(TA)6携带者的阿托伐他汀内酯水平更低(P<0.05)[12]。
-
阿托伐他汀易于口服吸收,其吸收取决于转运蛋白的活性,包括摄取性转运体(如有机阴离子转运多肽OATPs)和外排性转运体(乳腺癌耐药蛋白BCRP和P-gp)。阿托伐他汀通过OATP1B1摄取进入肝细胞,通过ABC转运体家族成员(BCRP、P-gp)排泄到胆汁。
-
有机阴离子转运多肽1B1(OATP1B1)表达于人肝细胞的窦状膜上,负责他汀类药物的肝脏摄取,SLCO1B1基因编码。SLCO1B1的SNP影响他汀类药物的体内分布和效能,目前研究最多的两个SNPs为SLCO1B1 521T>C(rs4149056)和388A>G(rs2306283)。
在携带SLCO1B1和ABCG2野生型等位基因的中国和日本受试者的瑞舒伐他汀、阿托伐他汀和辛伐他汀的AUC比白种人更高,其中,阿托伐他汀AUC0-∞分别比白种人高53%和69%[13]。SLCO1B1 c.521CC基因型患者的阿托伐他汀AUC0-48比TT基因型高144%(P<0.001),比TC基因型高61%(P=0.049)[14]。521C等位基因携带者的阿托伐他汀和2-羟基阿托伐他汀的平均Cmax、AUC0-24h和AUC0-∞显著高于521TT基因型组,平均CL值较521TT基因型受试者低,521C等位基因受试者的阿托伐他汀肌毒性风险增加[15]。SLCO1B1 521T>C在阿托伐他汀的临床试验中均可增高AUC,388A>G多态性对疗效影响不显著。
一项体外研究表明[16],SLCO1B1 521T>C下调质膜上OATP1B1的表达以减少阿托伐他汀转运进肝细胞。SLCO1B1 521T>C通过降低肝脏对阿托伐他汀的摄取使药物对HMG-COA还原酶的抑制作用减弱,同时血药浓度升高。SLCO1B1 521C(rs4149056)与辛伐他汀、阿托伐他汀、洛伐他汀和普伐他汀的降脂作用减弱有关[17]。然而,Prado等[18]研究表明,阿托伐他汀对智利高胆固醇血症受试者的降脂作用与SLCO1B1 rs4149056和rs2306283多态性无关(P>0.05),但388A>G rs2306283与阿托伐他汀治疗后更高的HDL-C相关(P=0.02)。现有的研究支持 SLCO1B1 521T>C基因多态性可增加阿托伐他汀的血药浓度,携带突变基因型的患者应适量减少用药剂量。
-
乳腺癌耐药蛋白(BCRP)由ABCG2基因编码,BCRP在肝脏、小肠、胆管等多种正常组织和癌细胞中均有表达。
ABCG2 c.421C>A在非洲人群中突变频率较低,但在白种人中突变频率为10%~14%,在日本或中国受试者中突变频率高达35%[19]。ABCG2 c.421C>A显著影响阿托伐他汀和瑞舒伐他汀的药动学,ABCG2在抑制这类他汀药物肠道吸收方面起到重要作用。c.421AA基因型携带者的瑞舒伐他汀AUC0-∞分别比c.421CA和c.421CC基因型高100%和144%。c.421AA基因型携带者的阿托伐他汀AUC0-∞比c.421CC高72%[1]。携带ABCG2 rs2622604的日本患者的阿托伐他汀口服生物利用度比非携带者增加了55%(95%CI:16%~131%)[20]。亚洲人群的ABCG2 c.421C>A突变率较高,具有变异等位基因的患者使用他汀类药物需减少剂量。
-
MDR1中的同义单核苷酸多态性C3435T和非同义单核苷酸多态性G2677T/A是药物处置和疗效差异的潜在因素。ABCB1 TTT/TTT基因型携带者的辛伐他汀酸AUC0-12h和阿托伐他汀AUC0-∞分别比CGC-CGC高60%和55%,且TTT/TTT型的阿托伐他汀的半衰期比CGC-CGC型长24%(P<0.05)[21]。ABCB1 rs1128503 (1236C>T)、rs2032582 (2677G>T/A)和rs1045642 (3435C>T)显著影响阿托伐他汀的AUC和阿托伐他汀治疗的降脂效果[22]。
ABCB1 C3435T与阿托伐他汀疗效下降相关。Kajinami等[23]发现 C3435T多态性与降LDL-C作用减弱相关,并且仅在女性中存在变异基因型携带者升HDL-C作用增强的现象。Rodrigues等[24]评估了G2677T/A和C3435T MDR1多态性对阿托伐他汀治疗前后血脂水平的影响,阿托伐他汀疗效与MDR1多态性之间没有显著相关性(P>0.05)。单倍型分析显示,与非T/T基因携带者相比,T/T携带者有较高的基础TC和LDL-C水平,提示MDR1多态性可能对巴西高胆固醇血症患者的基础胆固醇水平有重要影响。DeGorter等[25]报道的一项病例对照研究中,未发现ABCB1 2677T与阿托伐他汀血药浓度之间的关联。由于ABCB1变异的临床研究结果不确定且不一致,目前不建议临床常规使用ABCB1基因分型来预测他汀类药物的毒性。尽管如此,ABCB1在他汀类药物转运中发挥重要作用,未来可能包括ABCB1变异的多基因指导他汀类药物治疗。
-
HMGCR是内源性胆固醇合成途径中的限速酶,他汀类药物竞争性抑制HMGCR。外显子13上选择性剪切HMGCR pre-mRNA,产生两个全长(FL)HMGCR和D13 HMGCR[26]。缺少外显子13的选择性剪接HMGCR转录物的表达量与体内TC、LDL-C、APOB和TC的降低呈负相关[27]。Yu等[28]的研究表明,HNRNPA1的过表达降低了HMGCR酶的活性,增强了LDL的摄取并增加APOB。同时,rs1920045是一种与HNRNPA1外显子8选择性剪接相关的SNP,其与他汀类药物降TC作用减弱有关。Leduc等[29]发现HMGCR的选择性剪接可以解释家族性高胆固醇血症患者对他汀类药物应答的22%~55%的差异。携带rs3846662 AA基因型女性在接受他汀治疗后LDL-C的降低百分比显著低于非AA基因型女性(38.4%vs46.2%,P<0.05)。rs3846662多态性和HMGCR mRNA的选择性剪接显著影响女性对他汀治疗的反应。Cuevas等[30]发现在接受阿托伐他汀治疗的智利患者中,HMGCR rs17671591 T等位基因比野生型C等位基因,显示出更明显地降低LDL-C和升高HDL-C效应(LDL降低8%,P=0.021;HDL增加7.7%,P=0.039)[22]。
-
载脂蛋白E (ApoE)是脂质代谢的重要组成部分。ApoE基因的多态性与血脂水平和他汀类药物的降脂反应有关。APOE ε2等位基因的男性携带者LDL-C的降低率比野生型个体高7%~10% (P=0.01)[31]。Cerda等[32]报道APOE ε2等位基因者对高胆固醇血症具有保护作用,并减少致动脉粥样硬化的脂质分布。Deshmukh等[33]发现 APOE rs445925变异与阿托伐他汀治疗后LDL降低相关,而APOE rs4420638变异对阿托伐他汀治疗后LDL下降作用减弱相关。Jenny等[34]报道阿托伐他汀对139名智利受试者的降脂作用与APOE变异相关,E3/4基因型携带者的降胆固醇作用较E3/3基因型差(LDL-C:-18%vs-29%,P<0.001)。E2等位基因携带者的疗效优于E3和E4携带者,E4基因携带者需增加用药剂量才能达到目标调脂作用。
-
CYP7A1为胆固醇7a羟化酶,是催化胆固醇合成胆汁酸排泄途径的限速酶。CYP7A1基因是胆固醇排泄途径中最重要的调控基因,CYP7A1的基因突变可能会影响他汀类药物的反应。rs8192870位于CYP7A1基因第一个内含子,与阿托伐他汀降低LDL有关,阿托伐他汀治疗后,GG/GA基因型携带者LDL水平降低27.89%,AA基因型携带者LDL水平降低35.26%(P=0.021)[35]。
-
ABCG5/G8的胆固醇排泄和CYP7A1催化的胆汁酸生物合成是胆固醇排泄到胆汁中的主要途径。ABCG8 D19H变异体与阿托伐他汀治疗后降低LDL-C作用增强相关,而降TC作用没有显著性差异[36]。ABCG8 H19等位基因携带者比野生型D19等位基因携带者降低LDL-C作用更显著。ABCG8 H19和CYP7A1 C-204 等位基因对阿托伐他汀反应以剂量依赖性方式相互作用[37]。这些组合多态性分析比单一多态性分析(CYP7A1为4.2%,ABCG8 D19H为3.0%)更能有效解释降LDL-C差异,临床上应组合多态性分析以指导特定基因型患者合理用药。
-
胆固醇酯转运蛋白CETP虽然不是他汀类药物的药物靶点,但在胆固醇逆向转运过程、脂蛋白的分解代谢、不同脂蛋白类型的转化中发挥重要作用。CETP 629C/A的突变频率为0.412。与CC或CA基因型携带者相比,AA基因型携带者具有较低的CETP水平(P=0.026)和较高的HDL-C水平(P=0.035)。连续服用阿托伐他汀一年后,CC基因型携带者比CA或AA基因型降低LDL-C、降低LP(A)(P=0.005),升高HDL-C(P=0.045)疗效更显著(P<0.001)[38]。
-
他汀类药物是临床上首选的调血脂药物,并不是所有患者对他汀类药物反应良好,多数患者并未达到调脂目标值,还有部分患者出现不良反应(肌毒性、横纹肌溶解、肝转氨酶持续升高等)。
-
他汀相关肌肉症状(SAMS)是最常见的药物不良反应,常导致患者用药依从性差或停药。他汀类肌炎的特征是肌肉组织炎症导致肌肉疼痛或无力,并伴有肌酸激酶浓度持续升高。SAMS症状出现在开始他汀类药物治疗的最初6个月内,并在他汀类药物剂量降低或停止后消退,他汀类药物治疗需要密切监测血清肌酸激酶(CK)水平以预防SAMS。临床应用阿托伐他汀的群体中,SAMS的发病率更高。欧洲动脉粥样硬化协会共识小组发布的注册和观察性研究中,SAMS的发生率为7%~29%[39]。他汀诱导的肌毒性在体内外均呈现浓度依赖性,从轻度肌痛到潜在致命的横纹肌溶解或暴发性横纹肌溶解合并肌红蛋白尿最终导致急性肾功能衰竭。使用辛伐他汀的患者发生SAMS频率最高,其次是阿托伐他汀,氟伐他汀SAMS频率最低[40]。
遗传变异和高血药浓度是他汀类药物诱导肌病的关键原因。一项涉及1550名阿托伐他汀使用者的Meta分析表明,SLCO1B1 c.521T>C与阿托伐他汀不良反应之间存在显著相关性。未发现SLCO1B1 c.388A>G多态性与不良反应或疗效之间的关联(P>0.05)[41]。SLCO1B1 rs4149056与辛伐他汀治疗后肌病的发生显著相关,辛伐他汀诱导相关肌病的作用可能比阿托伐他汀更强[42]。与SLCO1B1 521 TT基因型携带者相比,TC或CC基因型阿托伐他汀的不良反应发生率高2.3倍[43]。Liu等[44]发现携带至少一个SLCO1B1 521C等位基因的患者发生肌毒性的风险更高。在接受瑞舒伐他汀治疗的个体中,SLCO1B1 521C突变等位基因突变与肌肉毒性风险显著相关。然而,521C突变等位基因与阿托伐他汀和辛伐他汀诱导肌毒性风险之间无显著相关性。SLCO1B1 c.521 T>C突变能降低OATP1B1的转运功能,使他汀类药物血药浓度升高,携带突变基因型的患者应降低药物剂量以减少不良反应。
CYP3A 基因型与阿托伐他汀诱导的肌肉损伤严重程度增加有关。在具有CYP3A5*3变异等位基因的个体中,肌酸激酶水平高于野生型个体(P=0.01)[45]。CYP3A基因型与阿托伐他汀诱导肌肉损伤的严重程度增加有关。服用阿托伐他汀的CYP3A5*3/*3基因型患者出现肌痛的可能性更大且更严重[45]。因此,携带CYP3A5*3变异等位基因的个体应减少药物剂量以减轻肌肉损伤程度。发生他汀类相关横纹肌溶解不良反应约60%与药物相互作用有关[46]。临床上使用CYP3A4酶的抑制剂或诱导剂会影响阿托伐他汀的血药浓度,导致发生不良反应的风险增加。
-
他汀类药物的另一种常见不良反应是肝毒性。肝毒性的发生率以血中转氨酶浓度升高为特征,远低于SAMS。ABCB1 rs2032582可预测日本人群患阿托伐他汀诱导的肝损伤(AILI)的风险。ABCB1中的一个SNP位点(rs2032582:2677G>T/A)与AILI显著相关(P=0.00068,OR=2.59,95%CI为1.49~4.50),提示G等位基因可能是AILI的危险因素。利用稳定表达ABCB1 rs2032582编码的ABCB1蛋白的Flp-In-293细胞,研究阿托伐他汀的细胞毒性,明确ABCB1 rs2032582 G等位基因在体内和体外是一个重要的AILI危险因素[47]。ABCB1 rs2032582可能是导致AILI的危险因素,携带该突变基因的个体应减少用药剂量。
-
阿托伐他汀的有关药物代谢酶、转运体、药物作用靶点以及脂质代谢相关基因多态性对阿托伐他汀药动学及药效学的影响,国内外已有很多研究,但仍不完善且许多研究结论不一致,可能是选取人群种族和样本数量的差异造成。目前缺乏多基因相互作用对他汀类药物治疗的影响研究,未来还需参考更多基因多态性以指导临床他汀类药物个体化合理用药。
Research progress of atorvastatin gene polymorphism
-
摘要: 阿托伐他汀是临床上广泛使用的调血脂药物,长期使用能够预防并减少动脉粥样硬化性心血管疾病(ASCVD)的发生,但阿托伐他汀的疗效具有显著的个体间差异,有些个体不能达到预期调脂目标值或出现严重的不良反应。这与个体间的遗传多样性有关,遗传变异可导致药物体内处置不同,从而导致临床疗效和不良反应有差异。对影响阿托伐他汀药物反应的药物代谢酶、药物转运体、药物作用靶点及与脂质代谢相关基因多态性加以综述,并从基因水平上探讨不同个体使用阿托伐他汀的药动学、药效学及不良反应易感性差别的原因。Abstract: Atorvastatin is a blood lipid-lowering drug widely used clinically. Long-term use can prevent and reduce the occurrence of atherosclerotic cardiovascular disease (ASCVD). However, the efficacy of atorvastatin has significant inter-individual differences. Some individuals failed to achieve the expected lipid-lowering target value or had serious adverse reactions, which were related to the genetic diversity between individuals. Genetic variation can lead to differences in drug configuration, clinical efficacy and adverse reactions. The drug metabolism enzymes, drug transporters, drug targets and genetic polymorphisms related to lipid metabolism were reviewed in this paper that affect the drug response of atorvastatin, and from the gene level to explore the reasons for the differences in the pharmacokinetics, pharmacodynamics and susceptibility to adverse reactions of different individuals using atorvastatin.
-
Key words:
- atorvastatin /
- blood lipids /
- drug metabolizing enzymes /
- drug transporters /
- gene polymorphisms
-
辅酶Q10(CoQ10)是生物体内广泛存在的脂溶性醌类化合物,作为细胞中的辅酶,其具有重要且广泛的药理作用[1],目前主要用于预防心血管疾病和增强免疫力等方面[2-4]。CoQ10分子量高、水溶性低,口服生物利用度低,注射剂稳定性差,对光十分敏感[5],越来越多的研究表明静脉注射乳剂是其理想载体[6]。
高油量可溶解更大量药物,对极难溶性药物的优势更为明显。但是目前对于含药静脉注射乳剂的研究中,油的质量体积比通常为10%[7],更高油量的研究相对较少。基于营养型脂肪乳剂的长期临床应用实践,高油量静脉注射乳剂的安全性已得到证实,具有广阔的发展空间和巨大的市场价值。同时乳剂不能经受冷冻也长期困扰着研究者们,卵磷脂与含亲水链段的乳化剂组合及恰当的油黏度是解决此问题的关键。本研究在乳剂经典处方中添加单唾液酸四己糖神经节苷脂(GM1),GM1含有一个较大的亲水头基,具有两亲性,利用其特殊的结构优势来解决乳剂不耐受冻融的难题。本研究旨在制备高油量、高载药量且耐冻融循环的CoQ10乳剂,建立其HPLC含量分析方法,并进行物理化学性质表征和稳定性评价,以弥补CoQ10现有制剂的不足,为高油量含药静脉注射乳剂的研究开发提供参考依据。
1. 仪器与试剂
BS124s电子分析天平(德国Sartorius公司);DF-101S 集热式恒温加热磁力搅拌器(巩义市英峪予华仪器厂);高速分散机(德国IKA T18 ULTRA TURRAX);M-110L型微射流仪(美国Microfluidics公司);垂直旋转自动高压灭菌器(沈阳天美达科学仪器有限公司);Nicomp-380 激光粒度测定仪(美国Particle Sizing Systems公司);PHS-2C型数字显示酸度计(上海伟业仪器厂);CS120GXL型超速离心机(日本Hitachi公司);UV228紫外-可见光检测器、P230高压恒流泵(大连依利特分析仪器有限公司);高精度全自动交流稳压器(浙江中川电气科技有限公司);AT-130柱温箱(天津市金洲科学仪器有限公司);人工气候箱(德国MMM公司)。
CoQ10(广东润和生物科技有限公司,批号:2017052405,纯度
$\geqslant $ 98%),中链油(MCT,辽宁铁岭北亚药用油有限公司,注射级),大豆油(LCT,辽宁铁岭北亚药用油有限公司,注射级),蛋黄卵磷脂(E80,德国Lipoid公司,注射级),GM1(重庆寰瑞生物制药有限公司,注射级),维生素E(VE,BASF维生素有限公司),甘油(湖南尔康制药有限公司,注射级),聚乙二醇12-羟基硬脂酸酯(HS15,德国BASF公司,注射级),葡萄糖注射液(昆明南疆制药有限公司),氯化钠注射液(吉林省都邦药业股份有限公司),灭菌注射用水(石家庄四药有限公司),其他试剂均为色谱纯。2. 方法与结果
2.1 CoQ10乳剂的制备
油种类对乳剂的稳定性至关重要,LCT和MCT单独应用时均存在各自的缺陷,且乳剂不能经受冷冻,因此将二者联合应用,质量比为1∶1[8]。结合静脉注射乳剂经典处方组成,CoQ10乳剂按如下方法制备[9]。称取质量体积比为30% LCT/MCT (质量比1∶1)、2% E80、0.05% VE和2% CoQ10于60 ℃水浴中加热熔融、分散均匀。2.25%甘油和0.2% GM1溶解于适量重蒸水中,60 ℃加热搅拌至完全溶解。待两相温度平衡后,高剪切搅拌下(8000 r/min)将水相加入油相中,完全加入后继续分散5 min,即得初乳。待初乳冷却至室温后,加重蒸水定容至处方量。将初乳转移至微射流仪中,经6 000 psi均质3个循环,14 000 psi均质6个循环减小粒径。依次过0.80和0.45 μm微孔滤膜。将所得乳剂分装,充氮气,121 ℃灭菌8 min。
2.2 CoQ10乳剂HPLC方法的建立
2.2.1 色谱条件[10]
色谱柱:Betasil C18(200 mm×4.6 mm,5 μm,大连依利特);流动相:甲醇-无水乙醇(体积比20∶80);柱温:35 ℃;流速:1.0 ml/min;检测波长:275 nm;进样量:20 μl。
2.2.2 溶液的配制
精密称定CoQ10 25.0 mg,置于25 ml量瓶中,用无水乙醇溶解并稀释至刻度,摇匀,即得浓度为1000.0 μg/ml的CoQ10对照品储备液。精密移取CoQ10乳剂0.1 ml,置于10 ml量瓶中,加入无水乙醇破坏乳剂并稀释至刻度,摇匀,过0.45 μm微孔滤膜,即得CoQ10乳剂样品溶液。
2.2.3 专属性试验
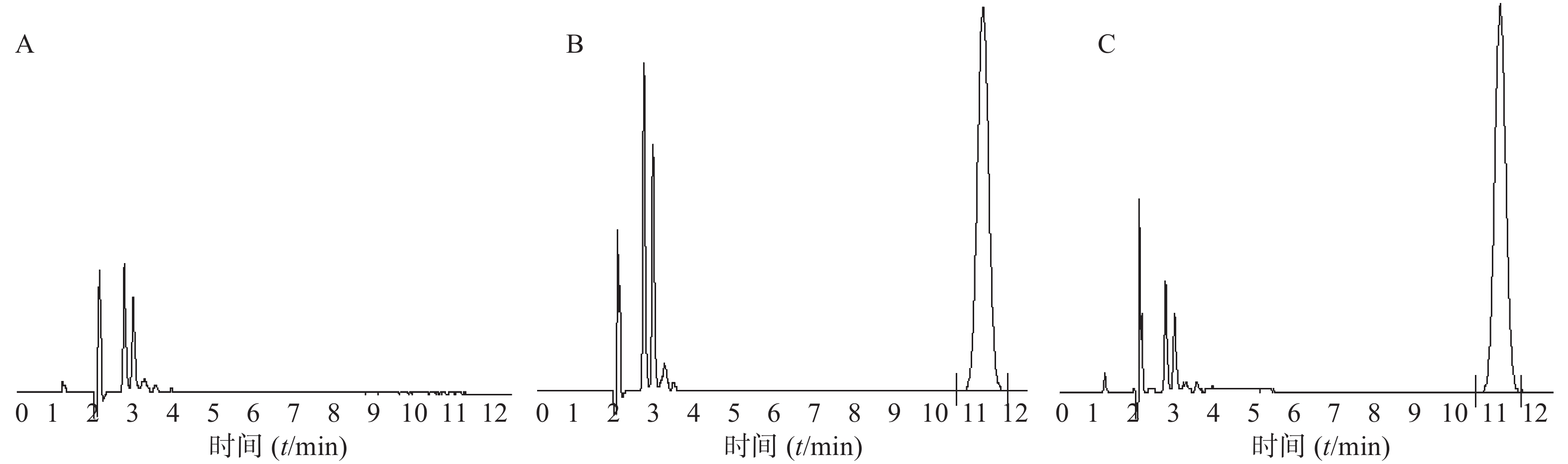
取CoQ10乙醇溶液20 μl进样,记录色谱图。将空白乳和CoQ10乳剂分别加适量无水乙醇破乳,经0.45 μm微孔滤膜滤过,取各续滤液20 μl进样,记录色谱图。在此色谱条件下辅料和溶剂对药物测定无干扰,见图1。
2.2.4 线性关系考察
精密量取CoQ10对照品储备液适量,用无水乙醇稀释成浓度为10.0、25.0、50.0、100.0、150.0、200.0、250.0 μg/ml的系列标准溶液,过0.45 μm微孔滤膜,各取20 μl进样分析,记录峰面积A。以A对浓度C(μg/ml)进行线性回归,得线性方程为A=15738 C- 12584,r=0.999 8。结果表明CoQ10在10.0 ~ 250.0 μg/ml范围内线性关系良好。
2.2.5 精密度试验
分别取低、中、高浓度(25.0、100.0、200.0 μg/ml)的CoQ10对照品溶液,于2、4、6、8、10 h各测定一次,以峰面积求算日内精密度;于1、2、3、4、5天各测定一次,以峰面积求算日间精密度。结果表明RSD均小于2%,方法精密度良好。
2.2.6 稳定性试验
取“2.2.2”项下CoQ10乳剂样品溶液室温下避光放置,于0、2、4、6、8、10、24 h分别取20 μl进样分析,记录峰面积。计算得各时间点峰面积为0 h测定值的100.45%、101.23%、99.27%、100.26%、99.34%及100.06%,表明样品溶液在24 h内稳定。
2.2.7 重复性试验
按“2.2.2”项下方法平行制备6份CoQ10乳剂样品溶液。另精密称定CoQ10适量,加乙醇溶解并稀释成浓度为200.0 μg/ml的溶液,摇匀,过0.45 μm微孔滤膜,即得对照品溶液。精密量取样品和对照品溶液20 μl进样,记录峰面积。外标法计算得样品中药物浓度分别为201.3、199.6、198.2、200.7、199.2、198.4 μg/ml,RSD为0.62%,表明该方法重复性良好。
2.2.8 回收率试验
精密移取CoQ10对照品储备液0.25、1.0和2.0 ml各3份于10 ml量瓶中,分别加入0.1 ml空白乳,再加乙醇破坏乳剂并稀释至刻度,摇匀,过0.45 μm微孔滤膜。将上述各溶液进样分析,记录峰面积,按“2.2.4”项下标准曲线方程求得药物浓度,计算回收率。结果表明25、100和200 μg/ml CoQ10样品溶液的平均回收率分别为100.04%、99.63%和100.49%,RSD分别为0.29%、0.80%和0.52%。
2.3 CoQ10乳剂的质量评价
2.3.1 外观
按照“2.1”项下的处方工艺制备CoQ10乳剂,观察其外观。CoQ10乳剂外观为淡黄色均匀乳状液体,无油滴、不溶性成分或块状团聚物,见图2。
2.3.2 物理化学性质
取CoQ10乳剂适量,用蒸馏水稀释至适宜浓度,采用激光散射粒径测定仪测定3批CoQ10乳剂的粒径、粒径分布和Zeta电位。测定波长为632.8 nm,测定角为90°,测定温度为25 ℃。结果显示3批样品的平均粒径为(239.5±0.8)nm,未见大于5 μm的粒子,符合静脉注射液的要求;粒径为(0.300±0.011)nm;Zeta电位为(−32.28±2.04)mV。同时测定了CoQ10乳剂的pH值为(5.86±0.02),符合注射剂质量要求。
2.3.3 含量测定
精密量取CoQ10 乳剂0.1 ml(含CoQ10约2 mg)于10 ml量瓶中,加乙醇溶解并稀释至刻度,摇匀,过0.45 μm微孔滤膜,取20 μl进样,记录色谱图。另取相同浓度的药物溶液,同法测定。外标法计算乳剂中CoQ10的含量,最终测得3批供试品的标示量百分比分别为99.18 %、101.50 %和101.08%,RSD为1.24%。
2.3.4 包封率测定
采用超速离心法测定CoQ10乳剂的包封率。乳剂经过超速离心后,油相、乳化层和水相彻底分开,将油相与乳化层中的药物量合并考虑,通过测定水相中的药物量来计算包封率。精密移取CoQ10乳剂0.1 ml至10 ml量瓶中,加入乙醇稀释至刻度,混匀,过0.45 μm微孔滤膜,取续滤液进行HPLC检测,外标法计算药物总量。移取CoQ10乳剂4 ml至超速离心管中,温度为4 ℃,以50 000 r/min离心2 h。精密移取下层液体1.0 ml至10 ml量瓶中,乙醇稀释至刻度,混匀,过0.45 μm微孔滤膜,取续滤液进行HPLC检测,外标法计算水相中药物含量。按上述方法测得3批CoQ10乳剂的平均包封率为(98.5%±1.1)%,表明药物绝大部分存在于制剂的油相和油水界面层中。
2.4 灭菌和冻融稳定性
将CoQ10乳剂分装于西林瓶,充氮气密封,121 ℃灭菌8 min。参考《化学药物稳定性研究技术指导原则》中涉及乳剂冻融试验的方法,将灭菌后样品在−20 ℃冷冻48 h,40 ℃放置48 h为一个循环,共循环3次,每完成一个循环测定粒径。结果显示3批CoQ10乳剂均具有良好的灭菌稳定性和冻融稳定性,见表1。
表 1 CoQ10乳剂的灭菌和冻融试验结果批次 粒径(l/nm) 灭菌前 灭菌后 冻融1次 冻融2次 冻融3次 1 241.4 239.6 255.7 263.0 271.2 2 238.5 240.0 252.6 265.9 272.3 3 240.2 238.7 259.0 263.5 269.1 2.5 稀释稳定性
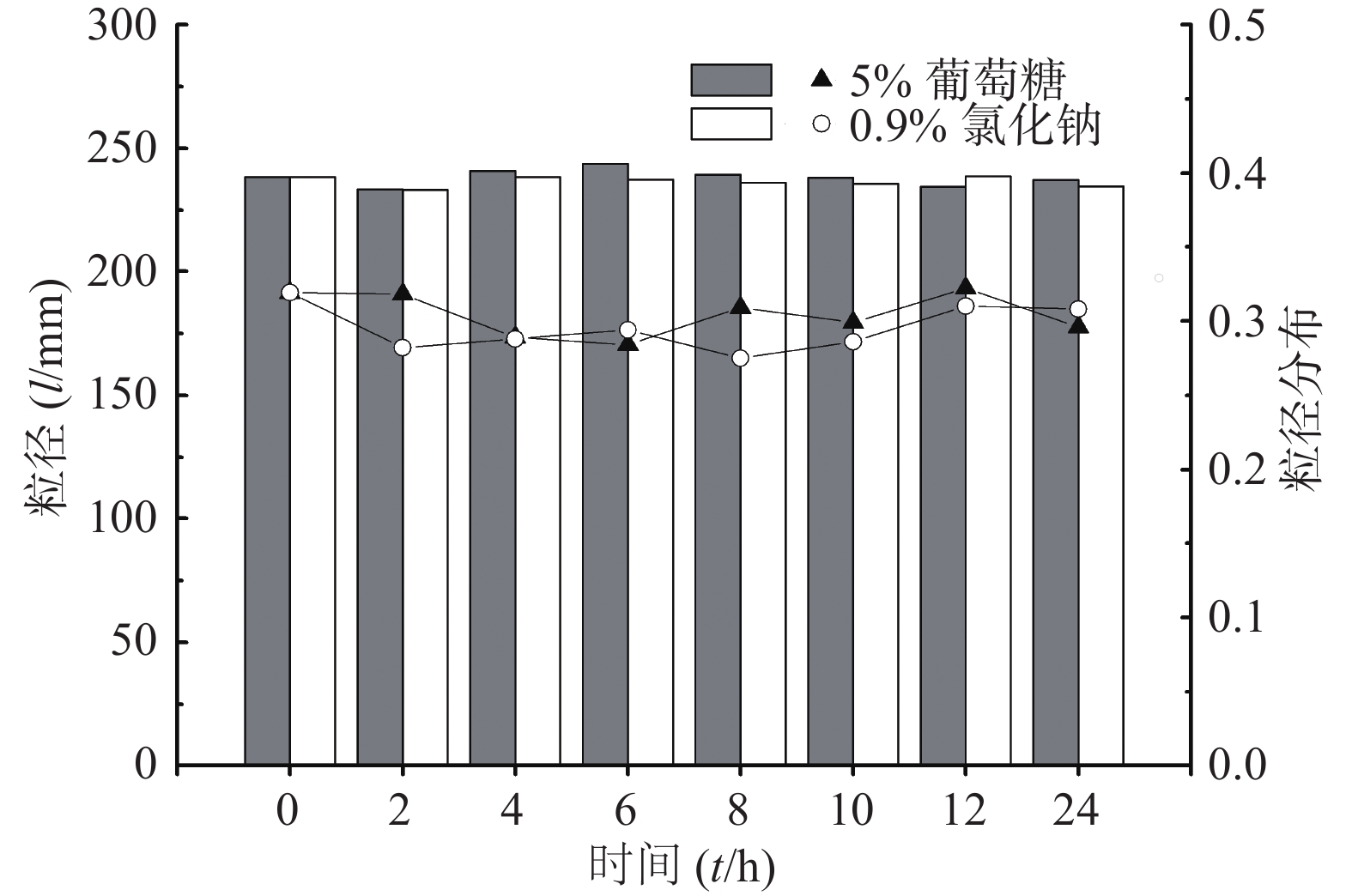
以平均粒径和粒径分布为指标,考察CoQ10乳剂在5%葡萄糖注射液和0.9%氯化钠注射液中的稀释稳定性。将制剂用上述稀释介质进行50倍稀释,于0、2、4、6、8、10、12、24 h取样,测定粒径和粒径分布。结果见图3,制剂经5%葡萄糖注射液和0.9%氯化钠注射液稀释后放置24 h,粒径和粒径分布均比较稳定,稀释稳定性良好。
2.6 光降解试验
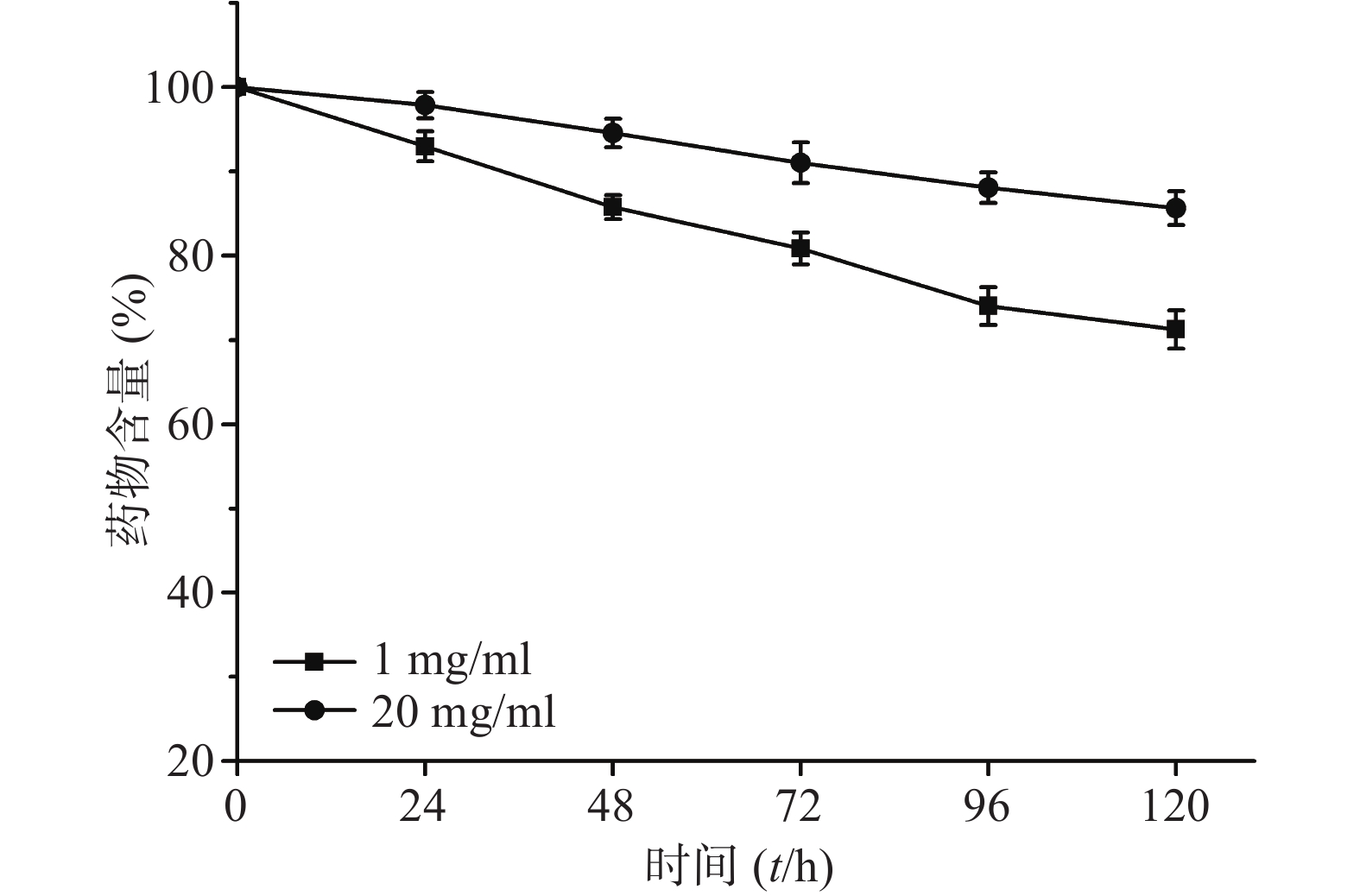
CoQ10结构中含有大量不饱和双键,是典型的光敏性药物。以药物含量为主要指标,考察不同因素对CoQ10乳剂光解速率的影响。将样品分装于透明西林瓶,置于人工气候箱中,于(25±2) ℃、(4500±500)lux条件下进行强制光解试验,调整样品与光源的距离约10 cm,使受光均匀,分别于0、24、48、72、96、120 h取样,HPLC法测定药物含量。绘制光降解曲线,并进行反应级数拟合,求出光解半衰期(t1/2)。
2.6.1 载体对光照稳定性的影响
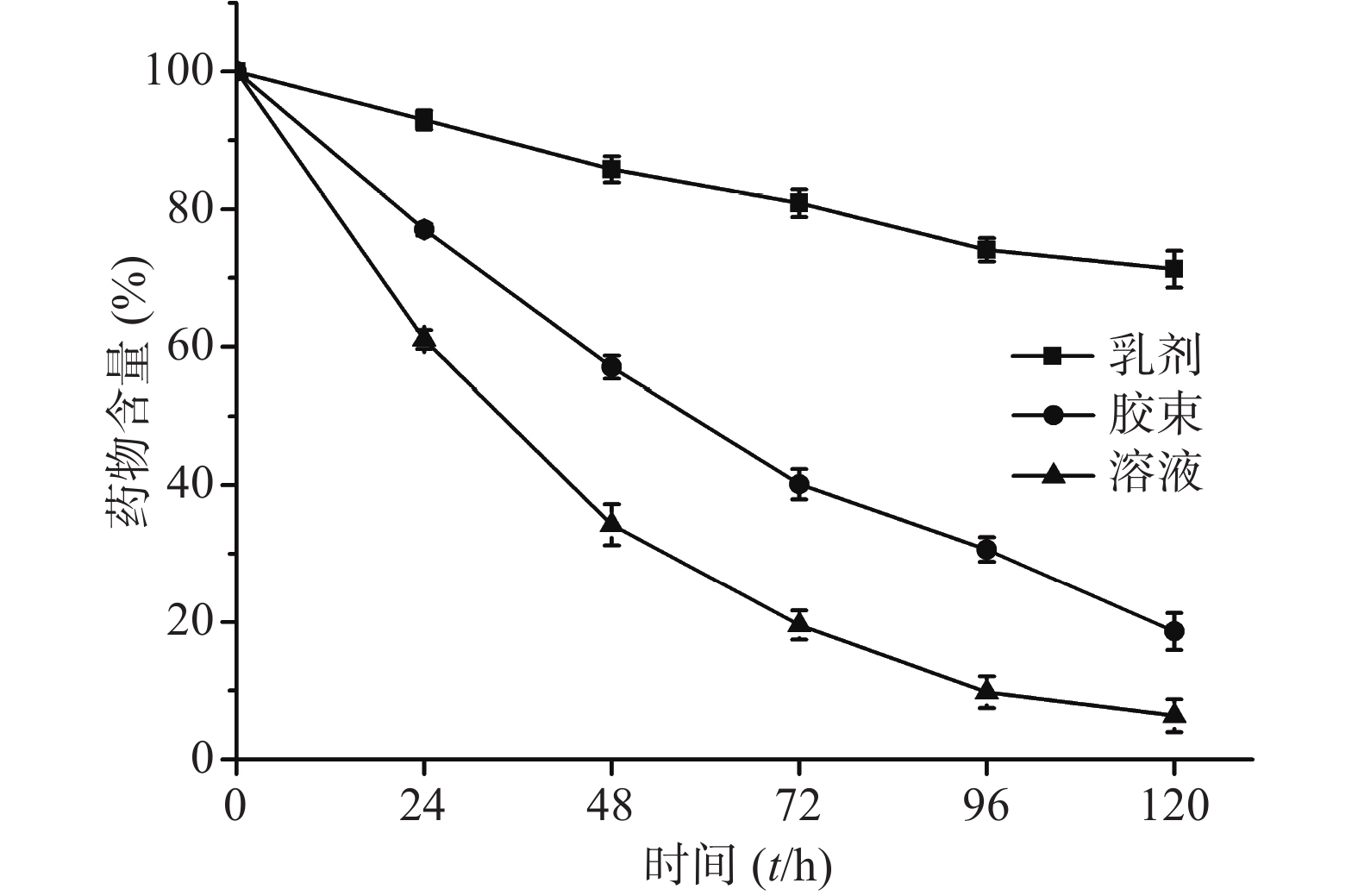
将适量CoQ10用无水乙醇溶解并稀释,即得CoQ10溶液。称取0.25% CoQ10和3% HS15于55 ℃水浴中加热熔融,分散均匀。搅拌状态下加入预热至相同温度的注射用水,搅拌10 min,即得CoQ10胶束。制备相同药物浓度(1 mg/ml)的CoQ10溶液、胶束和乳剂进行光解试验。光解曲线见图4,反应级数均为一级。对CoQ10的光保护作用由强到弱依次为乳剂、胶束和溶液,t1/2分别为239.0、50.6和29.4 h。
2.6.2 稀释倍数对光照稳定性的影响
将CoQ10乳剂(20 mg/ml)分别稀释0、2、4、10、20倍进行光解试验。光解曲线见图5,反应级数均为一级,稀释倍数由低到高,t1/2分别为533.1、495.0、288.8、115.5和62.4 h。
2.6.3 载药量对光照稳定性的影响
分别制备0.1%(1 mg/ml)和2.0%(20 mg/ml)载药量的CoQ10乳剂,进行光解试验。光解曲线见图6,反应级数均为一级,0.1%和2.0%载药量乳剂的t1/2分别为233.5和533.1 h。结果表明,光解速率与载药量呈反比,载药量越大,光解速率越小。
2.7 影响因素试验
CoQ10乳剂需在冰箱(2~8 ℃)内储存,因此以下稳定性实验参照《中国药典》2020版中对温度特别敏感药物制剂的实验条件进行。
2.7.1 光照稳定性
将CoQ10乳剂分装于棕色西林瓶中,充氮气、密封,在(25±2) ℃、(4 500±500) lux条件下,于第0、5、10天取样,以制剂外观、含量、粒径、pH为评价指标,考察CoQ10乳剂的光照稳定性。结果(见表2)表明3批制剂的含量和pH略有下降,提示该乳剂需避光制备与储存。
表 2 CoQ10乳剂的光照稳定性试验结果天数(t/d) 外观 含量(%) 粒径(l/nm) pH 0 良好 100.4 243.2 5.82 5 良好 97.7 239.8 5.79 10 良好 93.6 241.4 5.70 2.7.2 40 ℃高温稳定性
将CoQ10乳剂分装于西林瓶中,充氮气、密封,在(40±2) ℃条件下避光放置,于第0、5、10天取样,以制剂外观、含量、粒径、pH为评价指标,考察CoQ10乳剂在高温条件下的稳定性。结果(见表3)显示3批制剂的含量和pH随时间延长呈下降趋势。
表 3 CoQ10乳剂在40 ℃的稳定性试验结果天数(t/d) 外观 含量(%) 粒径(l/nm) pH 0 良好 100.4 243.2 5.82 5 良好 100.2 245.2 5.57 10 良好 99.5 245.0 5.21 2.8 25 ℃加速试验
取3批CoQ10乳剂分装于西林瓶中,充氮气、密封,在(25±2) ℃、相对湿度60%的条件下避光放置6个月,分别于第0、1、2、3、6个月取样,观察制剂外观,测定含量、粒径和pH值。结果表明,CoQ10乳剂在(25±2) ℃条件下避光放置6个月,各指标均在合格范围内,稳定性良好。
2.9 长期稳定性试验
取3批CoQ10乳剂分装于西林瓶中,充氮气、密封,在(5±3) ℃条件下避光放置12个月,分别于第0、3、6、9、12个月取样,观察制剂外观,测定含量、粒径和pH值。结果表明,CoQ10乳剂在(5±3) ℃条件下避光放置12个月,各指标均在合格范围内,稳定性良好。
3. 讨论
3.1 冻融稳定性
乳剂冷冻期间冰晶的形成会破坏乳化膜,从而引发小油滴聚集成大油滴,甚至导致浮油、破乳。解决乳剂冻融问题的主要手段为油相和乳化剂的合理选择。其一是油相要有恰当的黏度,将LCT与MCT联合应用既能克服单独使用存在的问题,又可取长补短,有利于乳剂冻融稳定性的提高。其二,牢固的乳化膜可增强乳剂的冻融稳定性,有研究[11]发现卵磷脂与含亲水链段的乳化剂组合对提高冻融稳定性有至关重要的作用。本实验CoQ10乳剂处方组成中的GM1是一种内源性糖脂,具有生物可降解、无免疫原性等特点[12],近年来已有研究将GM1应用于乳剂、脂质体和胶束的制备[12-14]。GM1是两亲性物质,可与卵磷脂形成复合乳化膜,增加膜的韧性。其结构中的较大亲水头基形成的空间立体位阻,及结构中唾液酸的荷负电对提高乳剂的冻融稳定性也至关重要。此外,在水相中加入含羟基的醇类或糖类(冻融保护剂),可以增加水相黏度,减慢冰晶的生长速度和程度,增强乳剂的冻融稳定性。
3.2 光照稳定性
CoQ10的光解具有浓度依赖性,起始浓度高时光解速率相对较慢,但当制剂稀释倍数增加时,其透光性大大增加,光解反应加剧。对于微粒给药系统,粒径是一个重要参数,同时其光解具有一定的粒径依赖性。本研究所制备的乳剂属于高油量低磷脂含量,与普通乳剂相比粒径较大,因此光稳定性要更好。推测原因为光不容易穿透大粒径制剂的内部,降低了药物的曝光率;此外,粒径大时粒子的曲率较低,比表面积小,因此曝光机会少,越不易光解[10]。本实验证明了将CoQ10包封入乳剂中可减少其曝光机会,对药物能起到一定的保护作用。但CoQ10乳剂仍需低温氮气环境下保存,使用过程中避光处理,以减少光解的概率。
-
[1] KESKITALO J E, ZOLK O, FROMM M F, et al. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin[J]. Clin Pharmacol Ther,2009,86(2):197-203. doi: 10.1038/clpt.2009.79 [2] KAJINAMI K, TAKEKOSHI N, BROUSSEAU M E, et al. Pharmacogenetics of HMG-CoA reductase inhibitors: exploring the potential for genotype-based individualization of coronary heart disease management[J]. Atherosclerosis,2004,177(2):219-234. doi: 10.1016/j.atherosclerosis.2004.09.004 [3] HE B X, SHI L, QIU J, et al. The effect of CYP3A4*1G allele on the pharmacokinetics of atorvastatin in Chinese Han patients with coronary heart disease[J]. J Clin Pharmacol,2014,54(4):462-467. doi: 10.1002/jcph.229 [4] GAO Y, ZHANG L R, FU Q. CYP3A4*1G polymorphism is associated with lipid-lowering efficacy of atorvastatin but not of simvastatin[J]. Eur J Clin Pharmacol,2008,64(9):877-882. doi: 10.1007/s00228-008-0502-x [5] WANG D, GUO Y, WRIGHTON S A, et al. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs[J]. Pharmacogenomics J,2011,11(4):274-286. doi: 10.1038/tpj.2010.28 [6] BECKER M L, VISSER L E, VAN SCHAIK R H, et al. Influence of genetic variation in CYP3A4 and ABCB1 on dose decrease or switching during simvastatin and atorvastatin therapy[J]. Pharmacoepidemiol Drug Saf,2010,19(1):75-81. doi: 10.1002/pds.1866 [7] KAJINAMI K, BROUSSEAU M E, ORDOVAS J M, et al. CYP3A4 genotypes and plasma lipoprotein levels before and after treatment with atorvastatin in primary hypercholesterolemia[J]. Am J Cardiol,2004,93(1):104-107. doi: 10.1016/j.amjcard.2003.08.078 [8] ROSALES A, ALVEAR M, CUEVAS A, et al. Identification of pharmacogenetic predictors of lipid-lowering response to atorvastatin in Chilean subjects with hypercholesterolemia[J]. Clinica Chimica Acta,2012,413(3-4):495-501. doi: 10.1016/j.cca.2011.11.003 [9] KIVISTÖ K T, NIEMI M, SCHAEFFELER E, et al. Lipid-lowering response to statins is affected by CYP3A5 polymorphism[J]. Pharmacogenetics,2004,14(8):523-525. doi: 10.1097/01.fpc.0000114762.78957.a5 [10] PENG C, DING Y, YI X, et al. Polymorphisms in CYP450 genes and the therapeutic effect of atorvastatin on ischemic stroke: a retrospective cohort study in Chinese population[J]. Clin Ther,2018,40(3):469-477.e2. doi: 10.1016/j.clinthera.2018.02.002 [11] CHO S K, OH E S, PARK K, et al. The UGT1A3*2 polymorphism affects atorvastatin lactonization and lipid-lowering effect in healthy volunteers[J]. Pharmacogenetics Genom,2012,22(8):598-605. doi: 10.1097/FPC.0b013e3283544085 [12] STORMO C, BOGSRUD M P, HERMANN M, et al. UGT1A1*28 is associated with decreased systemic exposure of atorvastatin lactone[J]. Mol Diagn Ther,2013,17(4):233-237. doi: 10.1007/s40291-013-0031-x [13] BIRMINGHAM B K, BUJAC S R, ELSBY R, et al. Impact of ABCG2 and SLCO1B1 polymorphisms on pharmacokinetics of rosuvastatin, atorvastatin and simvastatin acid in Caucasian and Asian subjects: a class effect? Eur J Clin Pharmacol,2015,71(3):341-355. doi: 10.1007/s00228-014-1801-z [14] PASANEN M K, FREDRIKSON H, NEUVONEN P J, et al. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin[J]. Clin Pharmacol Ther,2007,82(6):726-733. doi: 10.1038/sj.clpt.6100220 [15] WANG Y, TIAN Y, LV P, et al. The effect of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and 2-hydroxyatorvastatin in healthy Chinese people[J]. Die Pharmazie,2017,72(6):365-368. [16] KAMEYAMA Y, YAMASHITA K, KOBAYASHI K, et al. Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells[J]. Pharmacogenet Genomics,2005,15(7):513-522. doi: 10.1097/01.fpc.0000170913.73780.5f [17] PETERS B J M, RODIN A S, KLUNGEL O H, et al. Pharmacogenetic interactions between ABCB1 and SLCO1B1 tagging SNPs and the effectiveness of statins in the prevention of myocardial infarction[J]. Pharmacogenomics,2010,11(8):1065-1076. doi: 10.2217/pgs.10.81 [18] PRADO Y, SAAVEDRA N, ZAMBRANO T, et al. SLCO1B1 c. 388A>G polymorphism is associated with HDL-C levels in response to atorvastatin in Chilean individuals[J]. Int J Mol Sci,2015,16(9):20609-20619. doi: 10.3390/ijms160920609 [19] GRADHAND U, KIM R B. Pharmacogenomics of mrp transporters (ABCC1-5) and bcrp (ABCG2)[J]. Drug Metab Rev,2008,40(2):317-354. doi: 10.1080/03602530801952617 [20] TSAMANDOURAS N, GUO Y Y, WENDLING T, et al. Modelling of atorvastatin pharmacokinetics and the identification of the effect of a BCRP polymorphism in the Japanese population[J]. Pharmacogenetics Genom,2017,27(1):27-38. doi: 10.1097/FPC.0000000000000252 [21] KESKITALO J E, KURKINEN K J, NEUVONEN P J, et al. ABCB1 haplotypes differentially affect the pharmacokinetics of the acid and lactone forms of simvastatin and atorvastatin[J]. Clin Pharmacol Ther,2008,84(4):457-461. doi: 10.1038/clpt.2008.25 [22] PODURI A, KHULLAR M, BAHL A, et al. Common variants of HMGCR, CETP, APOAI, ABCB1, CYP3A4, and CYP7A1 genes as predictors of lipid-lowering response to atorvastatin therapy[J]. DNA Cell Biol,2010,29(10):629-637. doi: 10.1089/dna.2009.1008 [23] KAJINAMI K, BROUSSEAU M E, ORDOVAS J M, et al. Polymorphisms in the multidrug resistance-1 (MDR1) gene influence the response to atorvastatin treatment in a gender-specific manner[J]. Am J Cardiol,2004,93(8):1046-1050. doi: 10.1016/j.amjcard.2004.01.014 [24] RODRIGUES A C, REBECCHI I M M, BERTOLAMI M C, et al. High baseline serum total and LDL cholesterol levels are associated with MDR1 haplotypes in Brazilian hypercholesterolemic individuals of European descent[J]. Braz J Med Biol Res,2005,38(9):1389-1397. doi: 10.1590/S0100-879X2005000900014 [25] DEGORTER M K, TIRONA R G, SCHWARZ U I, et al. Clinical and pharmacogenetic predictors of circulating atorvastatin and rosuvastatin concentrations in routine clinical care[J]. Circ Cardiovasc Genet,2013,6(4):400-408. doi: 10.1161/CIRCGENETICS.113.000099 [26] JOHNSON J M, CASTLE J, GARRETT-ENGELE P, et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays[J]. Science,2003,302(5653):2141-2144. doi: 10.1126/science.1090100 [27] MEDINA M W, KRAUSS R M. The role of HMGCR alternative splicing in statin efficacy[J]. Trends Cardiovasc Med,2009,19(5):173-177. doi: 10.1016/j.tcm.2009.10.003 [28] YU C Y, THEUSCH E, LO K, et al. HNRNPA1 regulates HMGCR alternative splicing and modulates cellular cholesterol metabolism[J]. Hum Mol Genet,2014,23(2):319-332. doi: 10.1093/hmg/ddt422 [29] LEDUC V, BOURQUE L, POIRIER J, et al. Role of rs3846662 and HMGCR alternative splicing in statin efficacy and baseline lipid levels in familial hypercholesterolemia[J]. Pharmacogenet Genomics,2016,26(1):1-11. doi: 10.1097/FPC.0000000000000178 [30] CUEVAS A, FERNÁNDEZ C, FERRADA L, et al. HMGCR rs17671591 SNP determines lower plasma LDL-C after atorvastatin therapy in Chilean individuals[J]. Basic Clin Pharmacol Toxicol,2016,118(4):292-297. doi: 10.1111/bcpt.12493 [31] PEDRO-BOTET J, SCHAEFER E J, BAKKER-ARKEMA R G, et al. Apolipoprotein E genotype affects plasma lipid response to atorvastatin in a gender specific manner[J]. Atherosclerosis,2001,158(1):183-193. doi: 10.1016/S0021-9150(01)00410-5 [32] CERDA A, GENVIGIR F D, WILLRICH M A, et al. Apolipoprotein E mRNA expression in mononuclear cells from normolipidemic and hypercholesterolemic individuals treated with atorvastatin[J]. Lipids Health Dis,2011,10:206. doi: 10.1186/1476-511X-10-206 [33] DESHMUKH H A, COLHOUN H M, JOHNSON T, et al. Genome-wide association study of genetic determinants of LDL-c response to atorvastatin therapy: importance of Lp(a)[J]. J Lipid Res,2012,53(5):1000-1011. doi: 10.1194/jlr.P021113 [34] LAGOS J, ZAMBRANO T, ROSALES A, et al. APOE polymorphisms contribute to reduced atorvastatin response in Chilean Amerindian subjects[J]. Int J Mol Sci,2015,16(4):7890-7899. [35] JIANG X Y, ZHANG Q, CHEN P, et al. CYP7A1 polymorphism influences the LDL cholesterol-lowering response to atorvastatin[J]. J Clin Pharm Ther,2012,37(6):719-723. doi: 10.1111/j.1365-2710.2012.01372.x [36] KAJINAMI K, BROUSSEAU M E, NARTSUPHA C, et al. ATP binding cassette transporter G5 and G8 genotypes and plasma lipoprotein levels before and after treatment with atorvastatin[J]. J Lipid Res,2004,45(4):653-656. doi: 10.1194/jlr.M300278-JLR200 [37] KAJINAMI K, BROUSSEAU M E, ORDOVAS J M, et al. Interactions between common genetic polymorphisms in ABCG5/G8 and CYP7A1 on LDL cholesterol-lowering response to atorvastatin[J]. Atherosclerosis,2004,175(2):287-293. doi: 10.1016/j.atherosclerosis.2004.03.015 [38] GU G L, XU X L, YANG Q Y, et al. Effect of CETP polymorphism on atorvastatin lipid-regulating effect and clinical prognosis of patients with coronary heart disease[J]. Med Sci Monit,2014,20:2824-2829. doi: 10.12659/MSM.892711 [39] STROES E S, THOMPSON P D, CORSINI A, et al. Statin-associated muscle symptoms: impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management[J]. Eur Heart J,2015,36(17):1012-1022. doi: 10.1093/eurheartj/ehv043 [40] BRUCKERT E, HAYEM G, DEJAGER S, et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients: the PRIMO study[J]. Cardiovasc Drugs Ther,2005,19(6):403-414. doi: 10.1007/s10557-005-5686-z [41] DU Y M, WANG S Z, CHEN Z Y, et al. Association of SLCO1B1 polymorphisms and atorvastatin safety and efficacy: a meta-analysis[J]. Curr Pharm Des,2019,24(34):4044-4050. doi: 10.2174/1381612825666181219163534 [42] BRUNHAM L R, LANSBERG P J, ZHANG L, et al. Differential effect of the rs4149056 variant in SLCO1B1 on myopathy associated with simvastatin and atorvastatin[J]. Pharmacogenomics J,2012,12(3):233-237. doi: 10.1038/tpj.2010.92 [43] MIROŠEVIĆ SKVRCE N, MACOLIĆ ŠARINIĆ V, ŠIMIĆ I, et al. ABCG2 gene polymorphisms as risk factors for atorvastatin adverse reactions: a case–control study[J]. Pharmacogenomics,2015,16(8):803-815. doi: 10.2217/pgs.15.47 [44] LIU J E, LIU X Y, CHEN S, et al. SLCO1B1 521T > C polymorphism associated with rosuvastatin-induced myotoxicity in Chinese coronary artery disease patients: a nested case-control study[J]. Eur J Clin Pharmacol,2017,73(11):1409-1416. doi: 10.1007/s00228-017-2318-z [45] WILKE R A, MOORE J H, BURMESTER J K. Relative impact of CYP3A genotype and concomitant medication on the severity of atorvastatin-induced muscle damage[J]. Pharmacogenetics Genom,2005,15(6):415-421. doi: 10.1097/01213011-200506000-00007 [46] CHATZIZISIS Y S, KOSKINAS K C, MISIRLI G, et al. Risk factors and drug interactions predisposing to statin-induced myopathy: implications for risk assessment, prevention and treatment[J]. Drug Saf,2010,33(3):171-187. doi: 10.2165/11319380-000000000-00000 [47] FUKUNAGA K, NAKAGAWA H, ISHIKAWA T, et al. ABCB1 polymorphism is associated with atorvastatin-induced liver injury in Japanese population[J]. BMC Genet,2016,17(1):79. doi: 10.1186/s12863-016-0390-5 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 8050
- HTML全文浏览量: 3704
- PDF下载量: 73
- 被引次数: 0
