

 下载:
下载:



 下载:
下载:



-
水飞蓟素(SM)是从菊科植物水飞蓟Silybum marianum (L.)Gaertn.的果实中提取得到的一类天然的黄酮木脂素类成分,主要包含水溶性成分和脂溶性成分两大部分。其中,水溶性成分为二氢槲皮素,也称花旗松素(TF);脂溶性成分有水飞蓟宁(SD)、水飞蓟亭(SC)、水飞蓟宾(SB)和异水飞蓟宾(ISB)四种,水飞蓟宾和异水飞蓟宾为同分异构体且水飞蓟宾的含量较高,通常以水飞蓟宾对水飞蓟素进行质量控制[1-3]。现代药理学研究表明,水飞蓟素具有多种药理活性,除保护肝脏的功能之外[4],其对肿瘤、糖尿病、阿尔茨海默病等也有较好的疗效[5-7],是一种至关重要的药用原料。然而,水飞蓟素水溶性差导致其口服生物利用度很低,很大程度影响了其临床应用价值[8]。
微孔渗透泵(MPOP)近年来受到越来越多学者的关注。与传统渗透泵片相比,其省去了激光(或者机械)打孔的步骤,通过添加水溶性致孔辅料使得药物可以通过膜上的若干微孔释放出来,从而有效弥补了传统渗透泵工艺复杂的缺陷[9-10]。本研究前期以难溶性药物水飞蓟素为模型药物,通过磷脂复合物(PC)技术提高了水飞蓟素的溶出度,并进一步制备成水飞蓟素微孔渗透泵片(SM-PC MPOP),最终达到缓慢释药且提高生物利用度的目的。同时,以市售水飞蓟素胶囊为参比,对自制SM-PC MPOP体内的过程进行初步的探索,进而评价该制剂体外释放与体内吸收的相关性,为利用体外释放试验控制SM-PC MPOP的质量提供依据。
-
DHG-9145A型电热恒温鼓风干燥机(上海一恒科技有限公司);Agilent 1200型高效液相色谱仪(配备二极管阵列检测器,美国Agilent公司);RCZ-6BZ型药物溶出仪(上海黄海药检仪器有限公司);AL204型分析天平(Mettler Toledo仪器有限公司);Vortex-5型涡旋混合机(海门市Kylin-Bell有限公司);QB-212型摇摆式混匀器(海门市Kylin-Bell有限公司);LD5-2A型离心机(北京医用离心机厂)。
-
SM-PC MPOP片(自制,含SM 30mg,批号:131205);聚氧乙烯(阿拉丁试剂有限公司);氯化钠、醋酸纤维素、十二烷基硫酸钠(国药集团化学试剂有限公司);水飞蓟素胶囊(Legalon®,Madaus AG,规格:以SB计相当于SM140 mg);水飞蓟宾对照品(纯度:98.0%,上海同田生物技术股份有限公司,批号:120115);甲萘酚(国药集团化学试剂有限公司);β-葡萄糖醛酸酶(美国sigma公司);甲基叔丁基醚(色谱纯,上海阿拉丁试剂公司);其余试剂均为分析纯,水为蒸馏水。
-
健康成年比格犬,体质量(12±3) kg,购于四川省简阳市简城比尔动物养殖场,合格证号:SCXK(川)2009-15。
-
称取处方量的SM-PC(其制备工艺另文发表)、氯化钠、聚氧乙烯,等量递加法混合均匀,采用12 mm冲模直接压片(每片含SM 30 mg,硬度为40 N),即得微孔渗透泵的片芯。将醋酸纤维素、聚氧乙烯溶于丙酮-异丙醇(90∶10)中,即得包衣溶液。将片芯置于包衣锅内,温度为45 ℃,恒流泵流速3.0 r/min,压力0.5 MPa条件下进行包衣操作,直至衣膜达到预定要求为止。所得制剂在在40 ℃条件下干燥12 h,即得SM-PC MPOP片。
-
采用《中华人民共和国药典》溶出度测定法规定的桨法进行测定[11],以pH7.5的磷酸盐缓冲液900 ml为释放介质(添加0.5%十二烷基硫酸钠),温度(37±0.5) ℃,转速为100 r/min。于1、2、4、6、8、10和12 h各取出样品10 ml,用0.45 μm微孔滤膜滤过,及时补充等温度等体积的溶出介质10 ml。取续滤液进HPLC分析。另取SB对照品适量,精密称定,置于量瓶中,适量甲醇溶解并稀释至刻度,同法测定,计算不同时间内SM的主要成分SB的累积释放度,绘制释放曲线。
-
色谱柱为 Kromasil C18柱(4.6 mm×250 mm,5 μm),检测波长为288 nm,流速为1 ml/min,柱温40 ℃,进样量为20 μl,流动相为甲醇-0.05%磷酸的梯度洗脱(0~4 min,35% A;4~16 min,35% A→40% A; 16~23 min,40% A→45% A; 23~40 min,45% A→50% A)。
-
SB系列对照品溶液的制备:取SB对照品约10 mg,精密称定,置于25 ml量瓶中,加入适量的甲醇溶剂,轻微振摇使溶解,再添加一定量的甲醇稀释至刻度,配制得到浓度为464.0 μg/ml的SB对照品母液。用移液管精密移取该对照品母液0.25、0.5 ml于100 ml量瓶中;0.25、0.5、1、2和5 ml分别置于10 ml量瓶中,加65%甲醇溶液稀释至刻度,配成浓度依次为1.160、2.320、11.60、23.20、46.40、92.80和232.0 μg/ml的系列标准液,贮存备用。
甲萘酚内标溶液的制备:取甲萘酚约100 mg,精密称定,置于100 ml量瓶中,加一定量甲醇溶液,轻微振摇使溶解,再加适量的甲醇稀释至刻度,摇匀,精密吸取摇匀后的溶液5 ml于100 ml量瓶中,加65%甲醇稀释至刻度,制备得到浓度为54.80 μg/ml的内标液,贮存备用。
-
使用一次性采血针采集比格犬前肢的血液3.0 ml,置于真空采血管(添加肝素钠)中,用离心机在4 000 r/min的条件下离心10 min,取上层淡黄色液体,得到血浆样品。用移液枪精密吸取血浆(空白血浆或含药血浆)1.0 ml于10 ml具塞塑料离心管中,加入0.2 mol/L的磷酸盐缓冲液(pH 5.0)0.5 ml,用涡旋混合机混合均匀,再添加β-葡萄糖醛酸酶(酶活性相当于50 U)50 μl,涡旋混合30 s,37 ℃水浴中孵育约16 h,孵育结束后,添加内标液50 μl,混匀后加入pH 8.5的硼酸盐缓冲液2.0 ml,继续涡旋混合30 s,再添加甲基叔丁基醚4.0 ml,涡旋混合2 min使溶液均匀,最后置于3 000 r/min的离心机中离心10 min,分离得到上清液。将上清液置于40 ℃水浴下用氮气挥干,残渣用65%甲醇溶液100 μl复溶,涡旋混合1 min,精密吸取上清液20 μl进样。
-
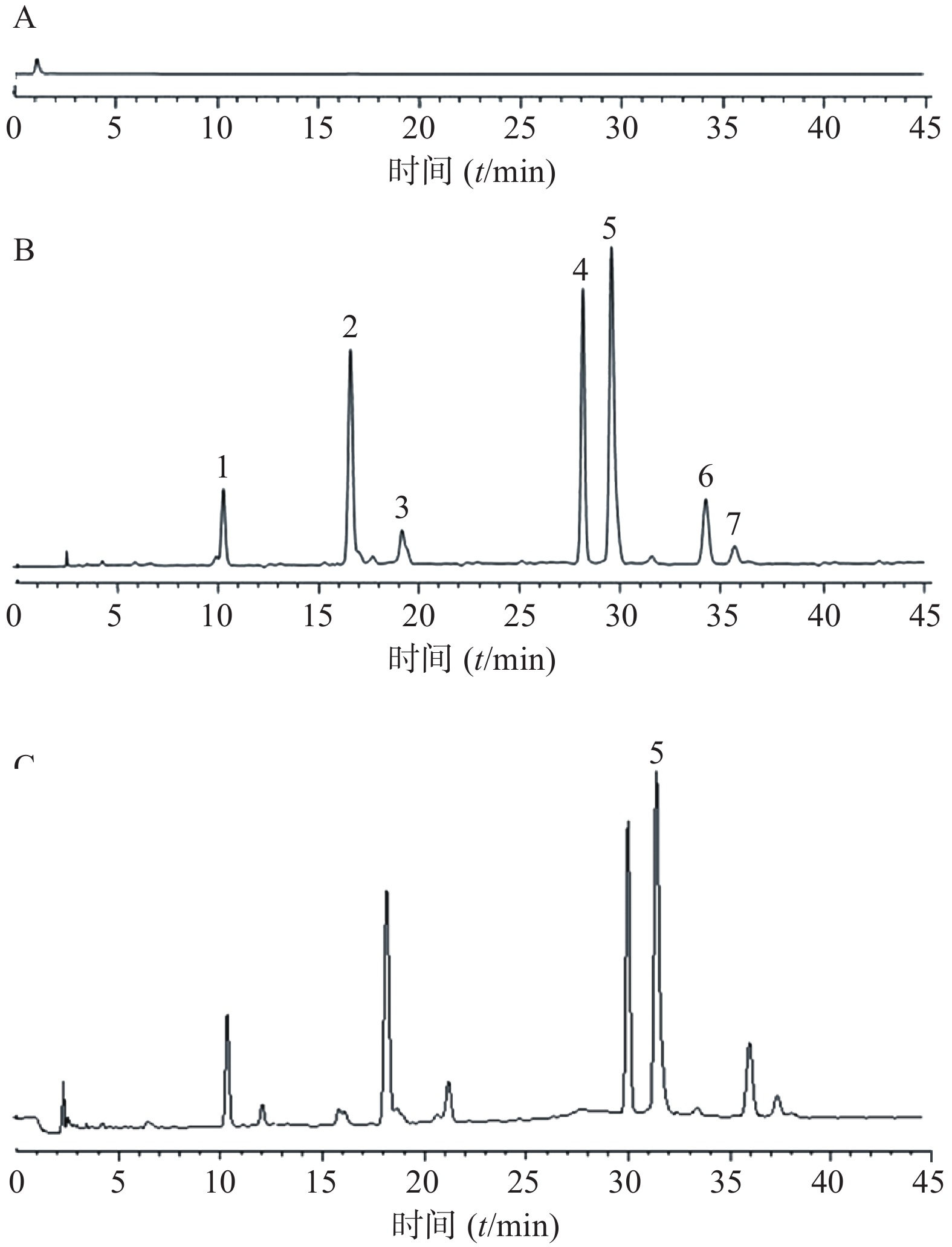
按照“2.3.1”项下色谱条件和“2.3.3”项下血浆样品处理方法处理和检测空白血浆、加药血浆和服药后血浆样品,记录色谱图,考察血浆内源性物质对内标、SB及其异构体的检测是否有干扰,结果见图1。
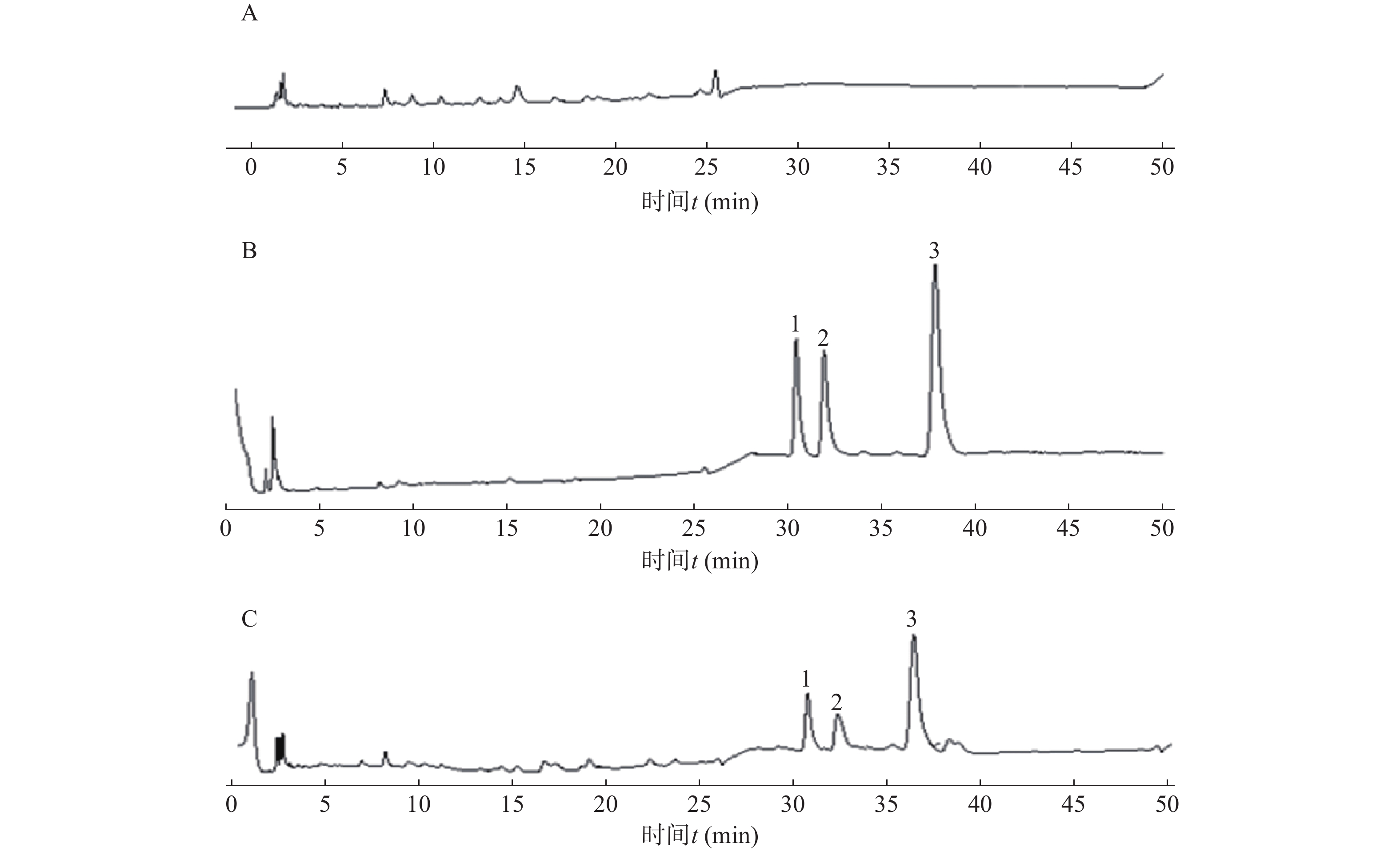
图 1 水飞蓟素血浆高效液相色谱图
由图1可见,SB、甲萘酚与血浆中的内源性物质分离良好,SB两个峰的保留时间分别为30.8 min和32.3 min,内标的保留时间为37.1 min。
-
精密吸取空白血浆1.0 ml,置于10 ml带塞圆底离心管,按“2.3.2”项下加入SB系列标准液50 μl,配制成浓度为0.060、0.120、0.580、1.160、2.320、4.640和11.60 μg/ml的血浆样品,按“2.3.3”项下方法处理,以SB的峰面积与内标甲萘酚峰面积的比值As/Ais对SB的浓度C(μg/ml)进行线性回归,用加权最小二乘法(权重为1/C)进行计算,得标准曲线方程:Y=0.680 7X+0.093 9(r=0.998 0),可见,在0.06~11.6 μg/ml的浓度范围内呈现良好的线性关系。
-
取空白血浆加入逐级稀释的SB标准溶液,按照信噪比为10的标准确定血浆样品中SB的最低定量限为0.05 μg/ml。
-
取低、中、高3个浓度(0.060、1.160、4.640 μg/ml)的血浆样品,按“2.3.3”项下方法处理,根据线性方程计算样品的浓度。于1天内连续测定5次,连续5天重复测定,计算日内、日间精密度和准确度,结果见表1。结果日内、日间精密度以及准确度均符合相关规定。
表 1 血浆样品中SB的精密度和准确度试验结果(n=5)
ρB
(μg/ml)日内试验 日间试验 $\bar x $±s
(μg/ml)RSD
(%)准确度
(%)$\bar x $±S
(μg/ml)RSD
(%)准确度
(%)0.060 0.060±0.006 12.56 88.3 0.051 8±0.004 8.12 86.3 1.160 1.072±0.084 7.84 92.4 1.084±0.077 7.07 93.4 4.640 4.240±0.252 5.95 91.4 4.368±0.028 8 6.60 94.1 -
取SB血浆样品(浓度为1.16 μg/ml),分别进行以下研究:(1)冻融考察:样品放置在冰箱中,冰冻温度为−80 ℃,解冻温度为25 ℃,反复冻融3次,进行稳定性考察。(2)长期冷冻考察:将SB血浆样品于−80 ℃的冰箱中放置1个月后进行稳定性考察。(3)血浆样品处理后24 h的稳定性考察:将血浆样品按“2.3.3”项下方法进行前处理,室温下保存24 h,按“2.3.1”项下条件进行测定,把测定的峰面积代入当天的标准曲线,计算回收率。结果表明,在3种稳定性考察中,SB的回收率>90.8%,RSD<9.5%(n=3),表明血浆样品反复3次冻融后稳定性良好;血浆样品在−80 ℃的冰箱中至少可以保存1个月;血浆样品处理后可在室温下保存24 h。因此,SB在比格犬血浆中的稳定性符合生物样品的检测要求。
-
取SB低、中、高3个浓度(0.06、1.16、4.64 μg/ml)的血浆样品,按“2.3.3”项下方法处理,进样测定SB与内标峰面积的比值A1,另取配制好的相应浓度的SB和内标溶液,取20 μl进样,得SB与内标峰面积比值 A2,按照公式:提取回收率(%)=(A1/A2)×100 %计算SB在低、中、高三个浓度的提取回收率为(68.59±8.37)%、(73.58±5.62)%、(70.26±4.15)%,同法测得内标甲萘酚的提取回收率为(81.8±3.51)%。
-
参考文献[12]并结合预实验,确定试验动物的给药剂量为水飞蓟素30 mg/kg。
-
采用双周期两制剂的双交叉实验设计,即将6只健康比格犬按体质量随机分成两组,交叉给予受试制剂(SM-PC MPOP)和参比制剂(市售水飞蓟素胶囊),洗脱期为 1 周。比格犬喂药前应先禁食12 h,空腹抽取空白血5 ml,按照“2.4.1”项下确定的剂量分别给予比格犬SM-PC MPOP及市售水飞蓟素胶囊,给药4 h以后进食低脂肪食物,在比格犬前肢小静脉处安置留置针,受试制剂组分别于0.25、0.5、0.75、1、1.5、2、3、4、6、8、10、12、24 h采血3 ml,参比试剂组分别于第0.5、1、1.5、2、3、4、6、8、10、12 h 各采血3 ml。 因受试制剂为缓释制剂,故在前1 h内采样点相较参比制剂(普通胶囊)密集。完成采样后间隔1周,1周后交叉给药,同时间点采血。血样置于用肝素抗凝的真空采血管中,立即于3 500 r/min的条件下离心取血浆,于−20 ℃保存,待测。
-
受试制剂和参比试剂的血药浓度-时间曲线见图2。
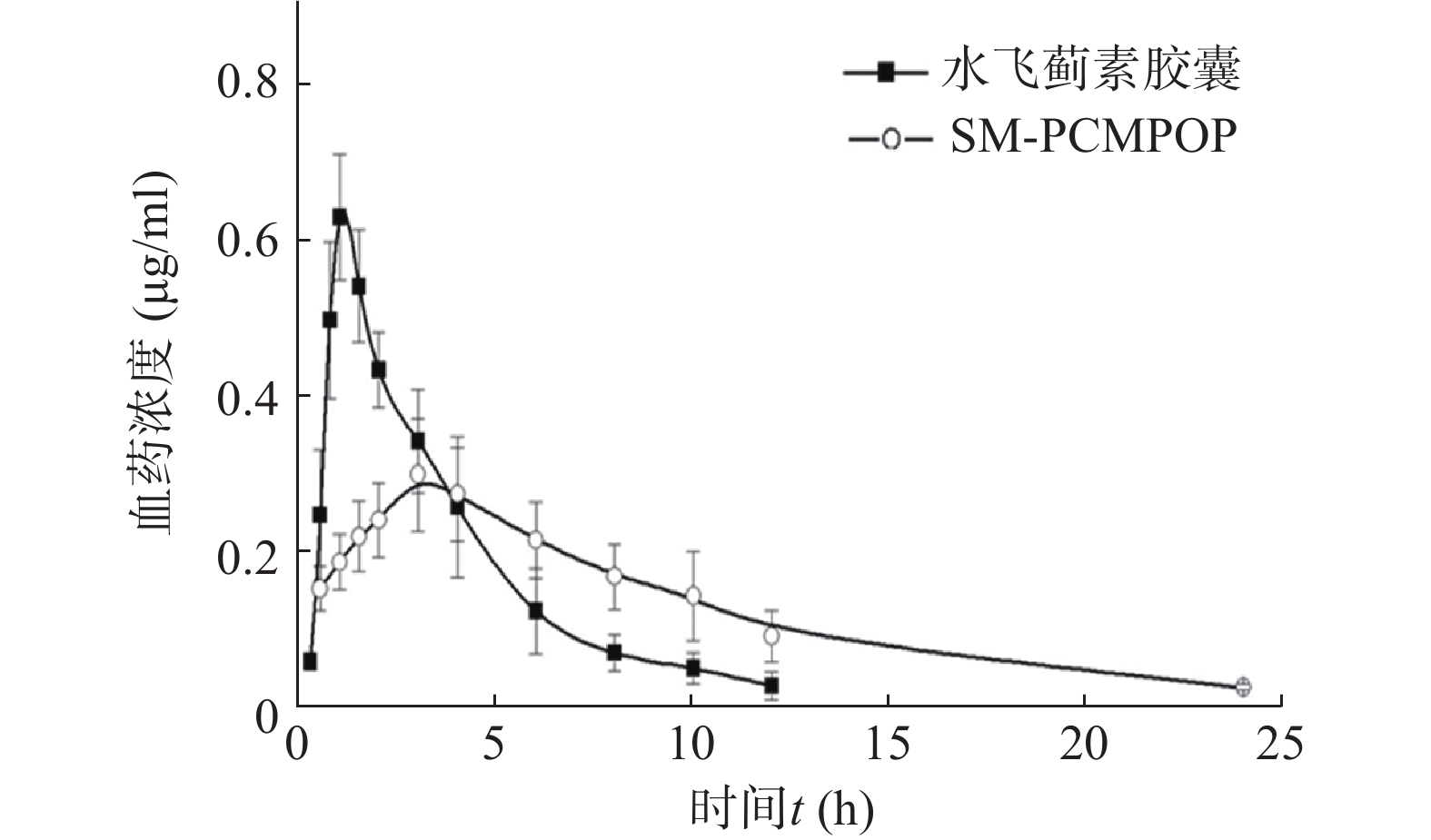
图 2 市售水飞蓟素胶囊与SM-PC MPOP的平均血药浓度-时间曲线(
$n=6 $ ) -
将受试制剂和参比制剂对应采血时间的血药浓度数据进行处理,计算非房室模型药动学参数,结果见表2。
表 2 非房室模型主要药动学参数(n=6)
药动学参数 单位 受试制剂 参比制剂 Tmax h 3.2±0.4 0.9±0.1 Cmax μg/ml 0.298 6±0.0689 0.629 9±0.076 5 ke h 0.119 4±0.017 6 0.274 2±0.100 5 t1/2 h 5.81±0.96 2.39±0.64 MRT h 3.469 4±0.075 8 7.485 7±0.082 4 AUC0→24 h·μg /ml 2.997 0±0.583 3 2.269 0±0.432 8 AUC0→∞ h·μg /ml 3.202 0±0.591 4 2.367 0±0.548 5 -
以非房室模型的数据计算SM-PC MPOP对市售水飞蓟素胶囊的相对生物利用度,可用下面的公式来计算:
$$ {F_r} = \frac{{{{(AU{C_{0 \to t}})}_T}}}{{{{(AU{C_{0 \to t}})}_R}}} \times 100\% $$ 虽受试制剂和参比制剂在剂量方面有所不同,但受试制剂具备线性药动学特征,故对计算结果进行剂量校正,计算得到Fr=(162.21±30.82)%。
-
反卷积分法(convolution)是FDA推荐的缓控释制剂评价体内外相关性的经典方法,其不需要进行房室模型拟合,直接用数学方法以真实的实验数据计算体内吸收分数。方程的实用形式如下:
$$ C(ti)={\displaystyle \sum _{k=1}^{i}Rk\cdot AU{C}_{ti-tk}^{ti-t_{K-1}}} $$ 由该方程可以解出:
$$ \begin{array}{l} R_1 = C(ti)/AUC_0^{t1} \\ R_2 = [C(t2) - R_1 AUC_{t2 - t1}^{t2}]/AUC_0^{t2 - t1} \\ R_3 = [C(t3) - R_1 AUC_{t3 - t1}^{t3} - R_2 AUC_{t3 - t2}^{t2}]/AUC_0^{t3 - t2} \\ R_i = [C(ti) - R_1 AUC_{ti - t1}^{ti} - R_2 AUC_{ti - t2}^{ti - t1} - R_{i-1}\\AUC_{ti - ti - 1}^{ti - ti - 2}]/AUC_0^{ti - ti - 1} \end{array} $$ 以市售水飞蓟素胶囊为参比,根据体内药动学试验结果计算出市售水飞蓟素胶囊的药-时曲线下面积及自制SM-PC MPOP各时间点的药物浓度,代入方程求出输入函数R(θ)。以R(θ)对相应时间点的累积释放百分率进行线性回归,直线的相关系数大于临界值表示体外释放与体内吸收相关性良好,结果见表3。
表 3 反卷积分参数
参数 时间(t/h) 2 4 6 8 10 12 SM-PC MPOP 释放度(%) 15.92 32.43 47.12 60.68 74.62 85.56 水飞蓟素胶囊分段AUC(h·μg/ml) 0.815 8 0.687 2 0.38 0.192 0.118 0.076 输入函数R(θ) 0.294 3 0.087 2 0.051 7 0.051 7 0.043 7 0.002 1 以累积释放百分率Y与输入函数R建立的回归方程为Y=−210.008 3R+71.301 7(r=0.839 0),当自由度df=n−2=4时,临界值r4,0.05=0.811,回归方程的相关系数r>r4,0.05,表明SM-PC MPOP的体外释放与体内吸收具有较好的相关性,即体外释放介质采用pH7.5的磷酸盐缓冲液(含0.5 %十二烷基硫酸钠)是可行的。
-
由于SM各组分中只有SB含量较高,其余成分含量均较低,采用HPLC法仅能测得主要成分SB的血药浓度。液质联用技术在近年来发展迅猛,不仅分离性能好,而且灵敏度高[13]。后续可借助超高效液相色谱-三重四级杆质谱联用技术(UPLC-MS/MS)同时检测SM的5种成分在比格犬血浆中的含量,更能科学地反映SM-PC MPOP各成分的体内吸收情况。
难溶性药物在血浆样品中多采用乙醚、甲基叔丁基醚、乙酸乙酯等进行萃取。试验中对这3种溶剂进行了考察,发现采用乙酸乙酯萃取时回收率较低,乙醚和甲基叔丁基醚无显著差异,但后者沸点比乙醚高,且在实际提取操作过程中溶剂挥发较少,安全性相对较高,因此选择甲基叔丁基醚作为萃取溶剂。
SB口服吸收以后,大部分在肝微粒体中经葡萄糖醛酸化结合形成苷,游离型含量较低,故加入β-葡萄糖醛酸酶水解苷键后测定血药浓度[14-15]。孵化前加入pH5.0的磷酸缓冲盐溶液调整血浆样品的 pH 以保证β-葡萄糖醛酸酶的活性,水解后再加入pH8.5硼酸盐缓冲液,使SB更多的以分子的形式存在,增加其脂溶性,再使用有机溶剂甲基叔丁基醚提取药物。其中,β-葡萄糖醛酸酶的加入量对测定结果准确性的影响至关重要,加入过少则难以水解完全,而加入过多会使具有紫外吸收的内源性物质被提取出来,增加干扰。
市售水飞蓟素胶囊(Legalon®)是一种高品质的水飞蓟素制剂,其采用特殊的工艺提取后,水飞蓟素纯度提高,水溶性增加[16],具有高含量、高纯度和高体外溶出度等特点。而自制的SM-PC MPOP则是基于卵磷脂复合技术,使得药物脂溶性增加,适宜的脂水分配系数使得药物更容易被胃肠道黏膜吸收,从而提高了生物利用度,两者在制剂技术方面的区别造成生物利用度有所差异。在本研究中,药动学研究结果显示自制的SM-PC MPOP峰浓度降低,实现了SM的成分缓慢释放的目标,且生物利用度高于市售水飞蓟素胶囊。
In vivo pharmacokinetics and in vitro-in vivo correlation of silymarin phospholipid complex microporous osmotic pump controlled-release tablets in beagle dogs
-
摘要:
目的 评价水飞蓟素磷脂复合物微孔渗透泵(SM-PC MPOP)控释片的体外释药特性、比格犬体内药动学及其体内外相关性。 方法 释放介质为pH7.5的磷酸盐缓冲液(添加0.5%十二烷基硫酸钠),以高效液相色谱法(HPLC)检测SM-PC MPOP的体外释放特征。用6只比格犬进行双周期交叉对照实验,按照30 mg/kg的剂量给药。HPLC法测定比格犬血浆内水飞蓟素的主要成分水飞蓟宾的质量浓度,应用药动学软件进行数据分析。 结果 SM-PC MPOP在12 h累积释放度超过85%。药动学研究情况表明,受试制剂(SM-PC MPOP)和参比制剂(市售水飞蓟素胶囊)在比格犬体内的主要药动学参数:Tmax分别为(3.2±0.4)、(0.9±0.1)h,Cmax分别为(0.298 6±0.068 9)、(0.629 9±0.076 5) μg/ml,AUC0→24分别为(2.996 8±0.583 3)、(2.268 9±0.432 8) h·μg /ml,SM-PC-MPOP对市售水飞蓟素胶囊的相对生物利用度为(162.21±30.82)%。 结论 自制的SM-PC MPOP实现了缓慢释药且增加生物利用度的目标,其体内吸收与体外释药具备相对较好的关联性(r=0.839 0)。 Abstract:Objective To evaluate the release characteristics in vitro, pharmacokinetics in rabbits and in vivo-in vitro correlation of silymarin phospholipid complex microporous osmotic pump controlled release tablets(SM-PC MPOP). Methods The release characteristics of SM-PC MPOP in vitro were detected by HPLC in the artificial gastric fluid. Six beagle dogs were subjected to double cycle cross control, which were given SM-PC MPOP and Legalon(30 mg/kg). The concentration of silybin in plasma was determined by HPLC and the data were processed by software. Results The cumulative release rate of SM-PC MPOP in vitro was over 85% in 12 h. The pharmacokinetics in beagle dogs showed that SM-PC MPOP and legalon conformed to double compartment first-order absorption model and the pharmacokinetic parameters were obtained: tmax:(3.2±0.4)and(0.9±0.1)h, Cmax:(0.298 6±0.068 9)and(0.629 9±0.076 5)μg/ml, AUC0→24:(2.996 8±0.583 3)and(2.268 9±0.432 8)h·μg /ml. The relative bioavailability of SM-PC MPOP was(162.21 ± 30.82)%. Conclusion SM-PC MPOP could release slowly, which could increase the relative bioavailability significantly. The correlation between the absorption in vivo and release in vitro was fine(r = 0.839 0). -
水飞蓟素(SM)是从菊科植物水飞蓟[Silybum marianum (L.) Gaertn.]的果实中提取的一种黄酮木脂素类混合物,主要成分为花旗松素(TF)、水飞蓟宁(SD)、水飞蓟亭(SC)、水飞蓟宾(SB)和异水飞蓟宾(ISB) [1-4]。现代药理学研究表明,水飞蓟素具有多种药理活性,除保肝护肝脏作用外[5],其对肿瘤、糖尿病、阿尔兹海默症(AD)也有一定的治疗效果[6-8]。然而,水飞蓟素在水中的溶解度低,导致其口服生物利用度达不到要求,极大地限制了其临床应用。
磷脂复合物(PC)是指药物和磷脂分子在非质子溶剂中反应,通过电荷迁移而形成稳定的化合物(或络合物),形成复合物后能显著改变母体药物的理化性质,进而提高药物的生物利用度[9]。当前,对天然药物磷脂复合物的研究已有较多报道,如根皮素磷脂复合物[10]、鼠李素磷脂复合物[11]、鞣花酸磷脂复合物[12]。然而,对SM-PC的制备工艺研究较少 [13],且针对其体外溶出行为研究主要以水飞蓟宾为指标进行评价,难以反映水飞蓟素原料药(SM-API)中多成分的情况。基于此,笔者拟采用星点设计-效应面法[10]优化SM-PC的制备工艺,并对其5种主要活性成分的体外溶出行为进行考察,以期解决水飞蓟素溶解性差、生物利用度低的问题,为后续水飞蓟素的进一步开发应用提供依据。
1. 仪器与试药
1.1 仪器
DHG-9145A型电热恒温鼓风干燥机(上海一恒科技有限公司);Agilent1200高效液相色谱系统(DAD检测器,美国Agilent公司);DZF-6051型真空干燥箱(上海一恒科技有限公司);AL204型分析天平(梅特勒-托利多仪器有限公司);RE-52A旋转蒸发仪(上海亚荣生化仪器厂);RCZ-6BZ型药物溶出仪(上海黄海药检仪器有限公司);KQ-800KDE型超声波清洗器(昆山市超声仪器有限公司);DK-S24型电热恒温水浴器(上海精密试验设备有限公司)。
1.2 试药
SM-API(辽宁盘锦华成制药厂,批号:120508,含量48 %);水飞蓟素胶囊(德国马博士制药厂,批号:B1101616,规格140 mg);TF、SC、SD、SB和ISB对照品(上海同田生物技术股份有限公司,纯度约为98.5%,批号:120115);大豆卵磷脂(江苏曼氏生物科技有限公司,批号:130201,其中,磷脂酰胆碱含量约为70 %);蛋黄卵磷脂(德国Lipoid®公司,批号:120920,其中,磷脂酰胆碱含量约为70 %);十二烷基硫酸钠(湖南尔康制药有限公司);甲醇为色谱纯,乙醇、丙酮、氯仿、磷酸、氢氧化钠等为分析纯,水为蒸馏水。
2. 方法与结果
2.1 SM-PC的制备及复合率的测定
取SM-API 5.0 g,加入丙酮溶液500 ml,充分搅拌使其溶解,加入大豆磷脂10.0 g,在40 ℃水浴条件下持续搅拌1 h,反应液减压蒸干,加入500 ml氯仿溶解,将溶液过滤,取续滤液减压浓缩至约50 ml,将浓缩液置于45 ℃的条件下真空干燥24 h,铲下得淡黄色磷脂复合物,粉碎过80目筛避光保存。根据SM-API不溶于氯仿,而SM-PC和PC均易溶解于氯仿,将制备的SM-PC溶解于氯仿当中,溶液用0.45 μm微孔滤膜过滤,收集沉淀,干燥称重,计算复合率,公式如下:
Y =(W−W1)/ W×100 %
其中,Y为复合率,W为SM-API的质量,W1为产生沉淀的质量。
2.2 分析方法的建立
2.2.1 色谱条件
Agilent1200高效液相色谱系统(DAD检测器,美国Agilent公司),色谱柱为Kromasil C18柱(4.6 mm ×250 mm ,5 μm),流速为1 ml/min,柱温40 ℃,检测波长为288 nm,进样量为20 μl,以甲醇(A)-0.05 %磷酸(B)为流动相,梯度洗脱(0~4 min,35% A;4~16 min,35% A→40 % A; 16~23 min,40% A→45% A; 23~40 min,45% A→50% A)[2]。
在上述色谱条件下,水飞蓟素5种成分的分离度均>1.5,结果见图1。
2.2.2 对照品溶液的制备
分别取TF、SC、SD、SB和ISB对照品适量,用分析天平精密称定,置于100 ml量瓶中,加甲醇轻微振摇使溶解,并稀释至刻度,得浓度为75.0、80.2、74.5、168.0、93.2 μg/ml的混合对照品溶液,保存备用。
2.2.3 供试品溶液的制备
取SM-PC适量(含水飞蓟素约20 mg),精密称定,置100 ml量瓶中,加甲醇轻微振摇使溶解,加65 %甲醇稀释至刻度,摇匀,0.45 μm微孔滤膜滤过,取续滤液作为供试品溶液。
2.2.4 线性关系及方法学考察
精密量取对照品混合液0.2、0.4、0.8、1.6、2.0、2.4、4.0 ml于10 ml量瓶中,加65 %甲醇稀释至刻度,配制成TF浓度为1.5、3.0、6.0、12.0、15.0、18.0、30.0 μg/ml;SC浓度为1.6、3.2、6.4、12.8、16.0、19.2、32.1 μg/ml;SD浓度为1.5、3.0、6.0、11.9、14.9、17.9、29.8 μg/ml;SB浓度为3.4、6.7、13.4、26.9、33.6、40.3、67.2 μg/ml;ISB浓度为1.9、3.7、7.5、14.9、18.6、22.4、37.3 μg/ml的混合对照品溶液,取混合对照品溶液20 μl进样,计算峰面积。以进样浓度X(μg/ml)为横坐标,峰面积Y为纵坐标,经线性回归得回归方程,结果见表1。
表 1 水飞蓟素5种有效成分的线性方程有效成分 线性方程 相关系数r 线性范围(μg/ml) TF Y=66.273X+3.921 0.9999 1.5~30.0 SC Y=42.952X−7.056 0.9999 1.6~32.1 SD Y=37.610 X+6.686 0.9998 1.5~29.8 SB Y=37.627X+6.684 0.9998 3.4~84.0 ISB Y=48.892X−4.681 0.9998 1.9~37.3 取SM-PC适量,按“2.2.3”项下方法平行制备 6 份供试品溶液,在“2.2.1”项下色谱条件进样测定,测得TF、SC、SD、SB和ISB峰面积RSD分别为0.86%、1.12%、0.65%、2.13%、1.87%,表明该方法重复性良好。 取同一份供试品溶液,于 0、2、4、8、12、24 h 在 “2.2.1”项下色谱条件进样测定,测得TF、SC、SD、SB和ISB峰面积RSD为 2.21%、0.98%、1.57%、2.76%、2.12%,表明溶液在 24 h 内稳定性良好。取“2.2.2”项下混合对照品溶液,在“2.2.1”项色谱条件下进样测定 6 次,测得TF、SC、SD、SB和ISB峰面积RSD分别为 0. 88%、0. 62%、0. 78%、1.05%、0.93%,表明仪器精密度良好。回收率试验结果表明,TF、SC、SD、SB和ISB的平均加样回收率分别为100.55%、99.64%、100.51%、100.13%、104.63%,RSD分别为0.95%、0.68%、1.15%、1.22%、1.05%。
2.3 SM-PC处方筛选的单因素考察
2.3.1 反应溶剂的影响
固体药物载体比1∶2,反应温度40 ℃,反应时间1 h,药物浓度为10 mg/ml,考察反应溶剂为无水乙醇、丙酮、甲醇时药物的复合率,结果见表2。
表 2 溶剂对复合率的影响(n=3)溶剂 介电常数 复合率(%) 无水乙醇 26.8 71.5±2.3 丙酮 19.5 75.6±2.0 甲醇 33.2 65.8±0.8 以丙酮为反应溶剂的复合率高于无水乙醇和甲醇,这可能是由于乙醇和甲醇均为质子性溶剂,不利于磷脂复合物的形成。
2.3.2 反应时间的影响
固定反应溶剂为丙酮,药物载体比1∶2、反应温度40 ℃,药物浓度为10 mg/ml,考察反应时间为0.5、1、2 、3 h时药物的复合率,结果见表3。
表 3 反应时间对复合率的影响(n=3)反应时间(t/h) 复合率(%) 0.5 46.1±1.0 1 76.4±1.4 2 85.1±1.2 3 86.6±0.5 4 87.3±1.2 试验表明,随着反应时间的延长,则复合率也增大,当反应时间大于2 h后,复合率受时间影响较小。
2.3.3 反应温度的影响
固定反应时间为2 h,反应溶剂为丙酮,药物载体比1∶2,药物浓度为10 mg/ml,考察磷脂复合物制备温度在30、40、50和60 ℃时的复合率,结果见表4。
表 4 反应温度对复合率的影响(n=3)温度(T/ ℃) 复合率(%) 30 68.2±0.8 40 86.7±2.2 50 90.8±0.8 60 92.4±1.3 试验表明,随着反应温度的升高,复合率也增大,在60 ℃时,复合率达到最高,考虑到磷脂在60 ℃以上时容易变性,因此未做更高温度的考察。
2.3.4 药物浓度的影响
固定反应温度为60 ℃,反应溶剂为丙酮,反应时间为2 h,药物载体比1∶2,考察药物浓度为5、10、20、30 mg/ml的复合率,结果见表5。
表 5 药物浓度对复合率的影响(n=3)药物浓度(ρB/mg·ml−1) 复合率(%) 5 92.5±1.2 10 91.5±2.2 20 88.7±0.8 30 75.8±1.3 从上表可见,药物浓度对药物的复合率有一定的影响,随着药物浓度的增加,水飞蓟素磷脂复合物的复合率降低。
2.3.5 水飞蓟素与大豆磷脂的比例
固定药物浓度为10 mg/ml,反应温度为60 ℃,反应溶剂为丙酮,反应时间为2 h,考察水飞蓟素原料药与大豆磷脂的比例分别为1∶0.5,1∶1,1∶1.5,1∶2,1∶2.5时药物的复合率,结果见表6。
表 6 反应物投料比例对复合率的影响(n=3)药物载体比(g/g) 复合率(%) 外观 1∶0.5 57.1±1.2 松脆 1∶1 83.0±2.2 松脆 1∶1.5 87.5±0.7 松脆 1∶2 92.2±1.2 松脆 1∶2.5 93.5±1.8 黏稠 由上表可以看出,随着磷脂用量的升高,复合率也增大,原因可能为磷脂的量增大,对水飞蓟素的增溶作用也增大,溶液分子间碰撞形成复合物的几率增大,但试验中我们发现,磷脂比例为1∶2.5时形成的复合物较黏稠,不易干燥,故选用药物载体比为1∶2。
2.3.6 磷脂的种类
固定反应温度为60 ℃,反应溶剂为丙酮,反应时间为2 h,药物浓度为10 mg/ml,水飞蓟素与磷脂的比例为1:2,考察载体采用蛋黄卵磷脂和大豆卵磷时对复合率的影响,结果见表7。
表 7 磷脂种类对复合率的影响(n=3)磷脂种类 磷磷脂酰胆碱含量(%) 复合率(%) 外观 大豆卵磷脂 70 91.8±0.3 松脆 蛋黄卵磷脂 70 92.2±0.2 黏稠 由上表可以看出,采用大豆卵磷脂和蛋黄卵磷脂作为载体时,其复合率并没有显著区别,但采用蛋黄卵磷脂制备得到的SM-PC较为黏稠,不易干燥,因此选用大豆卵磷脂作为复合载体。
2.4 星点设计-效应面法优化处方
2.4.1 试验设计
在单因素考察的基础上,选择对复合率影响较大的几个因素为自变量,即脂药比(X1)、反应浓度(X2)、反应温度(X3),以复合率(Y1)为因变量,采用三因素、五水平的星点试验设计,各因素的水平代码和具体的试验操作值见表8,具体的试验安排见表9。
表 8 各因素水平代码及试验操作值因素 −1.682 −1 0 +1 +1.682 脂药比 1 1.20 1.5 1.80 2 药物浓度(ρB/mg·ml−1) 5 8.04 12.5 16.96 20 反应温度(T/ ℃) 40 44.05 50.0 55.95 60 表 9 试验设计及测定结果(n=3)序号 X1 X2 X3 Y1 1 1.20 8.04 44.05 75.4±3.0 2 1.80 8.04 44.05 85.2±1.9 3 1.20 16.96 44.05 76.9±1.9 4 1.80 16.96 44.05 81.4±1.2 5 1.20 8.04 55.95 81.8±1.8 6 1.80 8.04 55.95 93.5±1.9 7 1.20 16.96 55.95 80.9±1.9 8 1.80 16.96 55.95 83.7±2.3 9 1.00 12.50 50.00 79.6±2.2 10 2.00 12.50 50.00 86.7±1.9 11 1.50 5.00 50.00 86.8±1.8 12 1.50 20.00 50.00 80.7±1.9 13 1.50 12.50 40.00 78.6±1.8 14 1.50 12.50 60.00 91.9±2.2 15 1.50 12.50 50.00 84.2±1.8 16 1.50 12.50 50.00 84.1±1.8 17 1.50 12.50 50.00 84.4±2.5 18 1.50 12.50 50.00 85.7±3.1 19 1.50 12.50 50.00 85.3±0.8 20 1.50 12.50 50.00 84.5±1.2 2.4.2 模型拟合
根据试验结果,应用SPSS 16.0统计软件,以评价指标(复合率)分别对各因素进行多元线性回归和二项式方程拟合,对二项式方程中的各系数进行t检验,删除P>0.2的项,以达到简化模型的目的。所得多元线性回归方程如下:Y=46.5846+10.034 2X1−0.381 9X2+0.534 1X3(r=0.8921),二项式方程如下:Y=75.3603+2.925 1X1+0.530 3X2−0.823 9X3−0.396 0X1X2+0.712 5 X1X3+7.039 1X2X3−7.180 2X12−0.0212 X22+2.845 9X32 (r=0.965 0)。
由以上方程可知,二项式方程拟合的回归系数r高于多元线性回归,故选择二项式拟合模型进行预测。
2.4.3 效应面优化与预测
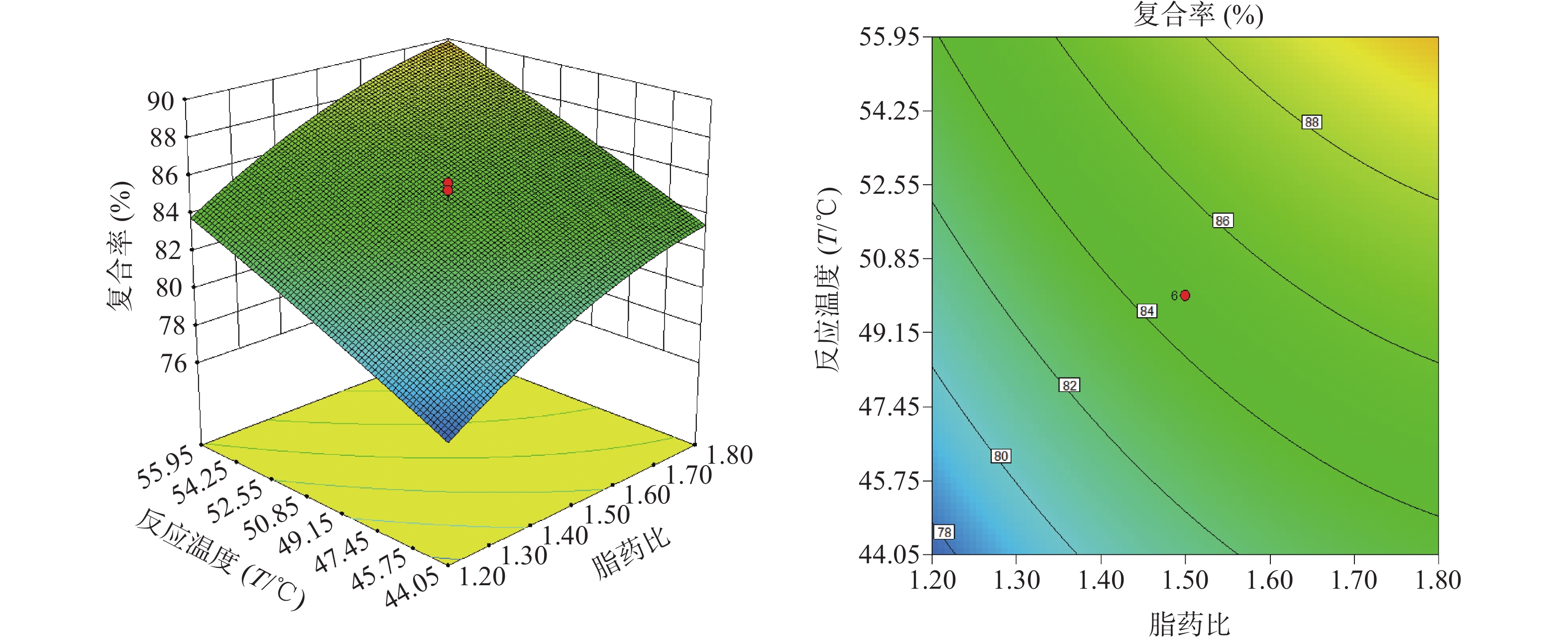
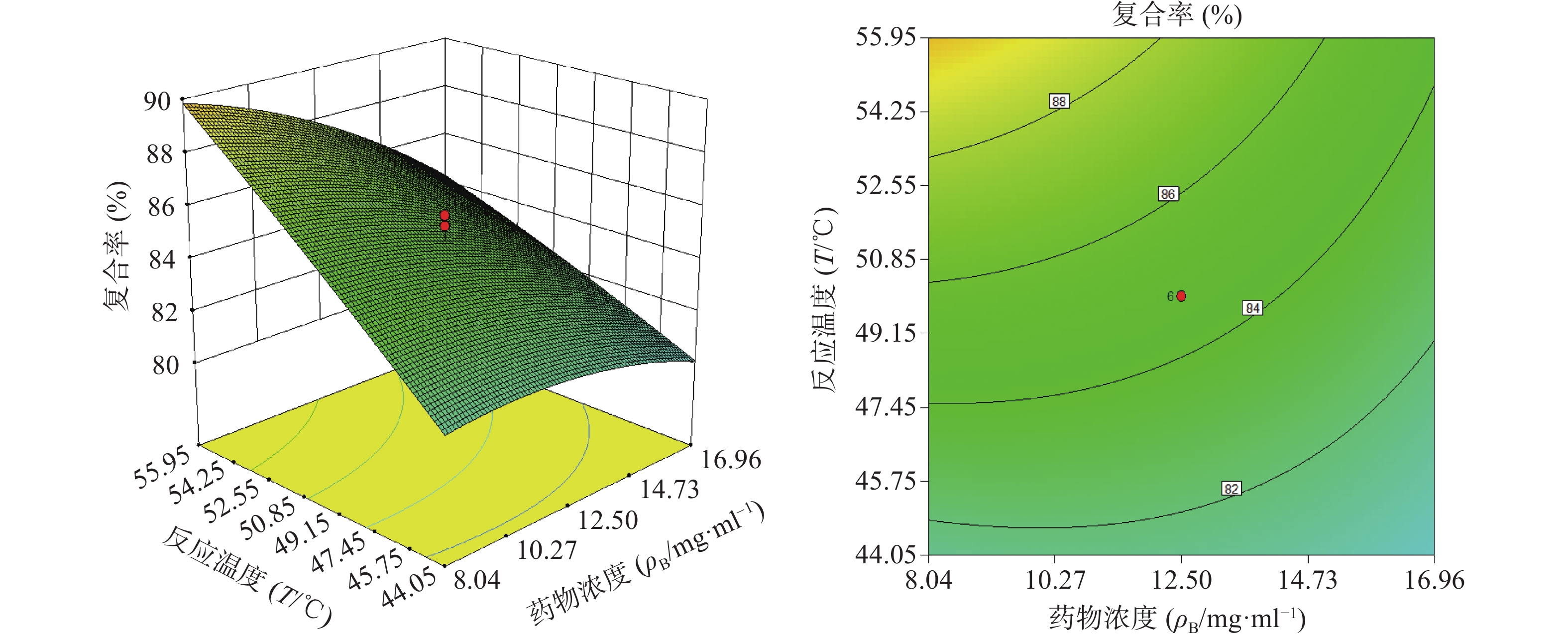
由“2.4.2”项下所得二项式方程,应用Design Expert 8.0软件绘制指标(复合率)与影响显著的两个自变量的三维效应面和二维等高图(另一个自变量设为中心点值),结果见图2、图3和图4。
由图2~4可以看出,在一定范围内,水飞蓟素与大豆卵磷脂的复合率随磷脂与水飞蓟素比例的增大而增高,随反应浓度的增高而降低,随反应温度的增高而增高。每个效应面都有其较优区域,以复合率为指标,软件给出最佳工艺是水飞蓟素与大豆卵磷脂比例为1∶1.8,反应温度55.95 ℃,反应物的浓度为8.04 mg/ml。综合考虑技术的可行性和经济的合理性,确定制备SM-PC的最优工艺条件为水飞蓟素与大豆卵磷脂的比例为1∶1.8,反应温度56 ℃,反应物的浓度8.0 mg/ml。
2.5 优化工艺的验证
将上述优选的最佳工艺制备3批SM-PC进行验证性试验,测定水飞蓟素与磷脂的复合率,将所得结果与二项式拟合方程的预测值相比较,结果见表10。
表 10 最佳工艺条件验证试验(%)批次 预测值 实测值 偏差 1 93.70 94.85 1.23 2 93.70 94.12 1.52 3 93.70 96.48 2.97 平均数 93.70 95.15 1.55 由表10可知,复合率的预测值和实测值的偏差小于3 %,证明试验所建立的数学模型预测性较好。
2.6 体外溶出试验
采用桨法测定SM-API、SM-PC、市售水飞蓟素胶囊在蒸馏水(pH5.6)和pH7.5的磷酸盐缓冲液(含0.5 % SDS)中的体外溶出度,桨转速为100 r/min,水浴温度为37 ℃。每次取含水飞蓟素150 mg的磷脂复合物粉末装入胶囊,分别于10、20、30、40、50、60 min 取样,每次10 ml,并及时补充等温度等体积的溶出介质,用0.45 μm微孔滤膜过滤后,取续滤液进样分析[14]。试验结果见图5和图6。SM-API、SM-PC和水飞蓟素胶囊在pH5.6的蒸馏水中溶出均不完全,SM-API各组分的溶出显示出较大差异,其中,水溶性成分TF溶出最高,达60%,而SB溶出不到20%。在pH7.5的磷酸盐缓冲液(含0.5 %SDS)中,SM-API的溶出度有了一定的提高,但仍溶出不完全,SM-PC各组分在60 min的溶出接近90%,主要成分SB的溶出行为与市售水飞蓟素胶囊相似,远高于原料药。
3. 讨论
在制备磷脂复合物的过程中,真空干燥必须控制一定的温度条件,温度太低,则磷脂复合物无法充分干燥,制备的磷脂复合物较黏稠,不利于后续制剂的制备;而温度太高,发现所制备的磷脂复合物颜色加深,这可能是由于大豆卵磷脂在高温条件下变性所致。结合预实验,本研究最终确定制备SM-PC的真空干燥温度为45 ℃。
通常选择溶出度试验的溶剂应该接近生理条件,对于难溶性药物而言, 可根据药物的理化性质(如溶解度数据)添加适量的表面活性剂。文献报道[15],将水飞蓟素制备成磷脂复合物后,药物的脂溶性提升较大,生物利用度在人体提高了2~ 3倍,而水溶性提升相对不明显,其在水、人工肠液和人工胃液中难以完全溶出。参考市售水飞蓟素磷脂复合物制剂的溶出条件,采用溶出介质为pH7.5的磷酸盐缓冲液(含0.5 %的SDS),桨转数为100 r/min[16]。
虽然采用磷脂复合物技术有效提高了水飞蓟素的体外溶出度,但由于磷脂复合物本身具有较强的黏性,分散性较差,因而也会限制其溶出速率和溶出度提高的幅度。从本文的研究结果也可以看出,仅采用磷脂复合物技术对水飞蓟素的体外溶出度提高有限,后续可与固体分散体、微球、微丸以及自微乳给药系统等制剂新技术联合应用来进一步改善溶出行为。需要注意的是,磷脂复合物联合其它制剂新技术虽能提高其自身的分散性,加快药物的溶出,但由于采用不同制剂新技术所制备药物的微粒载药方式、释药方式和速率、粒径大小等不同,其溶出速率、跨膜方式、作用部位也可能存在差异,也会影响药物的疗效[17]。因此,实际研究过程中,应该基于药物自身的性质以及循证药学的证据去探索合适的制剂新技术。
-
表 1 血浆样品中SB的精密度和准确度试验结果(n=5)
ρB
(μg/ml)日内试验 日间试验 $\bar x $ ±s
(μg/ml)RSD
(%)准确度
(%)$\bar x $ ±S
(μg/ml)RSD
(%)准确度
(%)0.060 0.060±0.006 12.56 88.3 0.051 8±0.004 8.12 86.3 1.160 1.072±0.084 7.84 92.4 1.084±0.077 7.07 93.4 4.640 4.240±0.252 5.95 91.4 4.368±0.028 8 6.60 94.1  下载: 导出CSV
下载: 导出CSV
表 2 非房室模型主要药动学参数(n=6)
药动学参数 单位 受试制剂 参比制剂 Tmax h 3.2±0.4 0.9±0.1 Cmax μg/ml 0.298 6±0.0689 0.629 9±0.076 5 ke h 0.119 4±0.017 6 0.274 2±0.100 5 t1/2 h 5.81±0.96 2.39±0.64 MRT h 3.469 4±0.075 8 7.485 7±0.082 4 AUC0→24 h·μg /ml 2.997 0±0.583 3 2.269 0±0.432 8 AUC0→∞ h·μg /ml 3.202 0±0.591 4 2.367 0±0.548 5
下载: 导出CSV
表 3 反卷积分参数
参数 时间(t/h) 2 4 6 8 10 12 SM-PC MPOP 释放度(%) 15.92 32.43 47.12 60.68 74.62 85.56 水飞蓟素胶囊分段AUC(h·μg/ml) 0.815 8 0.687 2 0.38 0.192 0.118 0.076 输入函数R(θ) 0.294 3 0.087 2 0.051 7 0.051 7 0.043 7 0.002 1
下载: 导出CSV
-
[1] THI T H, Nguyen, . Improving silymarin oral bioavailability using silica-installed redox nanoparticle to suppress inflammatory bowel disease[J]. J Control Release, 2021, 331: 515-524. doi: 10.1016/j.jconrel.2020.10.042 [2] OMMATI M M, FARSHAD O, AZARPIRA N, et al. Silymarin mitigates bile duct obstruction-induced cholemic nephropathy[J]. Naunyn-Schmiedeberg’s Arch Pharmacol, 2021, 394(6): 1301-1314. doi: 10.1007/s00210-020-02040-8 [3] ZENG Q P, LIU Z H, HUANG A W, et al. Preparation and characterization of silymarin synchronized-release microporous osmotic pump tablets[J]. Drug Des Devel Ther, 2016, 10: 519-531. [4] MASTRON J K, SIVEEN K S, SETHI G, et al. Silymarin and hepatocellular carcinoma: a systematic, comprehensive, and critical review[J]. Anticancer Drugs, 2015, 26(5): 475-486. doi: 10.1097/CAD.0000000000000211 [5] KIM S H, CHOO G S, YOO E S, et al. Silymarin induces inhibition of growth and apoptosis through modulation of the MAPK signaling pathway in AGS human gastric cancer cells[J]. Oncol Rep, 2019, 42(5): 1904-1914. [6] KUMAR J, PARK K C, AWASTHI A, et al. Silymarin extends lifespan and reduces proteotoxicity in C. elegans Alzheimer’s model[J]. CNS Neurol Disord Drug Targets, 2015, 14(2): 295-302. doi: 10.2174/1871527314666150116110212 [7] ATAIHIRE J U, NWANGWA E K, IGWEH J C. Modulations in anti-oxidant activities of selected gastro-intestinal tissues in alloxan-induced, silymarin treated diabetic wistar rats[J]. OJGas, 2019, 9(5): 73-90. doi: 10.4236/ojgas.2019.95010 [8] TVRDÝ V, POUROVÁ J, JIRKOVSKÝ E, et al. Systematic review of pharmacokinetics and potential pharmacokinetic interactions of flavonolignans from silymarin[J]. Med Res Rev, 2021, 41(4): 2195-2246. doi: 10.1002/med.21791 [9] 袁晓炫, 要辉, 张欣, 等. 天麻素微孔渗透泵缓释片的制备及其药动学评价[J]. 中国医院药学杂志, 2021, 41(17): 1742-1748. [10] 王玉华, 杨海燕, 郝海军. 环维黄杨星D包合物微孔渗透泵控释片的制备及处方优化[J]. 中成药, 2020, 42(10): 2555-2560. [11] 国家药典委员会. 中国药典2015年版四部[S]. 北京: 中国医药科技出版社, 2015: 121-124. [12] ZHU H J, BRINDA B J, CHAVIN K D, et al. An assessment of pharmacokinetics and antioxidant activity of free silymarin flavonolignans in healthy volunteers: a dose escalation study[J]. Drug Metab Dispos, 2013, 41(9): 1679-1685. doi: 10.1124/dmd.113.052423 [13] 王迪, 俞佳, 詹固, 等. 液质联用技术在中药研究中的应用进展[J]. 中华中医药学刊, 2022, 40(2): 68-71. [14] 李秉新, 王鑫, 刘有平, 等. 水飞蓟宾非对映异构体在大鼠肝微粒体中葡萄糖醛酸化代谢的酶动力学比较研究[J]. 沈阳药科大学学报, 2018, 35(4): 289-294, 305. [15] 肖衍宇, 宋赟梅, 陈志鹏, 等. 水飞蓟素前体脂质体的制备和大鼠药代动力学的研究[J]. 药学学报, 2005, 40(8): 758-763. [16] 周旖璇, 张纯刚, 尹丽, 等. 水飞蓟宾原料药、利加隆、水林佳大鼠体内药物动力学研究[J]. 亚太传统医药, 2021, 17(6): 11-15. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3155
- HTML全文浏览量: 1412
- PDF下载量: 16
- 被引次数: 0



