

 下载:
下载:
 下载:
下载: 

</td><td class="table_top_border" align="center" valign="middle">肾功能衰竭组(<i>n</i>=5)</td><td class="table_top_border" align="center" valign="middle">肾功能正常组(<i>n</i>=19)</td><td class="table_top_border" align="center" valign="middle"><i>P</i></td><td class="table_top_border" align="center" valign="middle">检验值</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">年龄(岁)</td><td class="table_top_border2" align="center" valign="middle">54.7±9.3</td><td class="table_top_border2" align="center" valign="middle">59.2±6.1</td><td class="table_top_border2" align="center" valign="middle">53.5±9.8</td><td class="table_top_border2" style="align-point:0pt;eft;" align="center" valign="middle">0.23</td><td class="table_top_border2" style="align-point:0pt;eft;" align="center" valign="middle">1.53</td></tr><tr><td align="center" valign="middle">体表面积(m<sup>2</sup>)</td><td align="center" valign="middle">1.6 (1.5, 1.6)</td><td align="center" valign="middle">1.4 (1.4, 1.5)<sup>▲</sup></td><td align="center" valign="middle">1.6 (1.5, 1.6)</td><td align="center" valign="middle">0.05</td><td align="center" valign="middle">3.86</td></tr><tr><td align="center" valign="middle">给药剂量(mg/m<sup>2</sup>)</td><td align="center" valign="middle">73.6(72.3,80.0)</td><td align="center" valign="middle">70.4 (69.4, 73.0)</td><td align="center" valign="middle">74.4(72.3, 91.2)</td><td align="center" valign="middle">0.08</td><td align="center" valign="middle">3.06</td></tr><tr><td align="center" valign="middle">体重(kg)</td><td align="center" valign="middle">51.5±5.1</td><td align="center" valign="middle">51.4±3.8</td><td align="center" valign="middle">51.5±5.5</td><td align="center" valign="middle">0.97</td><td align="center" valign="middle">0.001</td></tr><tr><td align="center" valign="middle">白蛋白(g/L)</td><td align="center" valign="middle">42.4±3.8</td><td align="center" valign="middle">39.0±4.9<sup>▲</sup></td><td align="center" valign="middle">43.2±3.0</td><td align="center" valign="middle">0.02</td><td align="center" valign="middle">6.12</td></tr><tr><td align="center" valign="middle">谷草转氨酶(U/L)</td><td align="center" valign="middle">24.6 (19.9, 31.7)</td><td align="center" valign="middle">19.3 (19.1, 20.6)<sup>▲</sup></td><td align="center" valign="middle">29.0(24.1, 34.9)</td><td align="center" valign="middle">0.01</td><td align="center" valign="middle">6.02</td></tr><tr><td align="center" valign="middle">谷丙转氨酶(U/L)</td><td align="center" valign="middle">21.4(15.0, 34.1)</td><td align="center" valign="middle">13.7(11.2, 14.8)<sup>▲▲</sup></td><td align="center" valign="middle">24.0(20.4, 39.4)</td><td align="center" valign="middle">0.002</td><td align="center" valign="middle">10.01</td></tr><tr><td align="center" valign="middle">总胆红素(μmol/L)</td><td align="center" valign="middle">11.0±3.7</td><td align="center" valign="middle">9.0±2.9</td><td align="center" valign="middle">11.5±3.7</td><td align="center" valign="middle">0.17</td><td align="center" valign="middle">1.95</td></tr><tr><td align="center" valign="middle">直接胆红素(μmol/L)</td><td align="center" valign="middle">2.4±1.1</td><td align="center" valign="middle">1.9±0.8</td><td align="center" valign="middle">2.5±1.1</td><td align="center" valign="middle">0.27</td><td align="center" valign="middle">1.28</td></tr><tr><td align="center" valign="middle">肌酐(μmol/L)</td><td align="center" valign="middle">60.5 (52.0, 285.8)</td><td align="center" valign="middle">908.0 (819.0, 1 018.0)<sup>▲▲</sup></td><td align="center" valign="middle">54.8 (52.0, 65.0)</td><td align="center" valign="middle">< 0.001</td><td align="center" valign="middle">11.42</td></tr><tr><td align="center" valign="middle">肌酐清除率(ml/min)</td><td align="center" valign="middle">69.3(17.9, 91.8)</td><td align="center" valign="middle">4.9(4.3, 5.4)<sup>▲▲</sup></td><td align="center" valign="middle">86.3(59.3, 92.5)</td><td align="center" valign="middle">< 0.001</td><td align="center" valign="middle">11.4</td></tr><tr><td align="center" valign="middle">化疗前白细胞(×10<sup>9</sup>/L)</td><td align="center" valign="middle">6.5±1.7</td><td align="center" valign="middle">7.5±1.7</td><td align="center" valign="middle">6.2±1.6</td><td align="center" valign="middle">0.12</td><td align="center" valign="middle">2.56</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">化疗前中性粒绝对值(×10<sup>9</sup>/L)</td><td class="table_bottom_border" align="center" valign="middle">4.0(3.0, 4.9)</td><td class="table_bottom_border" align="center" valign="middle">4.1(4.0, 8.7)</td><td class="table_bottom_border" align="center" valign="middle">3.9 (2.5, 4.8)</td><td class="table_bottom_border" align="center" valign="middle">0.21</td><td class="table_bottom_border" align="center" valign="middle">1.55</td></tr></tbody>
<tfoot><tr><td colspan="6" align="left" valign="middle"><sup>▲</sup><i>P</i><0.05,<sup> ▲▲</sup><i>P</i><0.01,与肾功能正常组比较。</td></tr></tfoot>
</table></div></foreignObject></svg>)
-
近年来由于糖尿病肾病、高血压肾病和癌症发病率的增加,肿瘤科医生会面对同时患有癌症和肾衰竭的患者[1]。乳腺癌是女性常见的恶性肿瘤,而化疗是目前乳腺癌治疗的主要方法之一。紫杉类药物多西他赛是乳腺癌化疗中的主要细胞毒性药物,它通过干扰细胞有丝分裂和有丝分裂间期所必需的微管网络发挥抗肿瘤作用[2]。而多西他赛的骨髓抑制、神经毒性等不良反应会影响其用量,甚至药物的选择。目前指南均推荐多西他赛按照体表面积进行给药[3],然而由于不同人群的基因多态性、生理状态、遗传特性,给予相同剂量的多西他赛仍会导致显著的药动学差异[4]。现临床上已广泛应用治疗药物血药浓度监测(TDM)来降低药物的个体差异[5]。已有大量研究表明[5–12],药时曲线下面积(AUC)为评价多西他赛体内暴露的药动学参数,并作为多西他赛剂量调整的依据。而在肾衰竭合并乳腺癌的患者中,多西他赛在剂量调整和透析对其的影响信息非常少[1,13],仍需开展进一步的研究。本研究旨在分析慢性肾衰竭对乳腺癌患者多西他赛的AUC及不良反应是否有影响,以便对此类患者的多西他赛给药剂量提供依据。
-
收集2019年1月至2021年11月我院收治的24例已行乳腺癌根治术的使用多柔比星注射液联合环磷酰胺4个周期序贯多西他赛4个周期(AC-T)辅助化疗的乳腺癌患者的临床资料及随访结果。本研究为单中心、回顾性、病例对照研究。根据患者的肾功能指标肌酐清除率(Ccr),研究分为慢性肾功能衰竭组和肾功能正常组:Ccr≤10 ml/min为慢性肾功能衰竭组,Ccr≥90 ml/min肾功能正常组。本研究经我院医学伦理委员会审核批准(KY2021-004-02)。
-
病例纳入标准:(1)年龄18岁以上;(2)经组织病理学确诊为乳腺癌;(3)使用AC-T辅助化疗;(4)器官水平满足以下要求:白细胞计数≥3.0×109/L,中性粒细胞绝对计数≥1.5×109/L,血小板计数≥75×109/L,血红蛋白≥90 g/L,天冬氨酸氨基转移酶、丙氨酸氨基转移酶低于正常值上限的2.5倍,血清总胆红素低于正常值上限的1.5倍;(5)监测了多西他赛血药浓度。排除标准:肝、心功能和血常规异常。
-
多西他赛注射液(齐鲁制药有限公司,批号1E0074C49 国药准字H20031244,规格 0.5ml∶20mg×1支/盒),使用AC化疗4个周期后使用,21天一次,治疗过程中如果病人出现不良反应且无法耐受时,可将药物进行减量,下调20%~25%[14]。
-
收集患者一般资料:性别、年龄、体重、体表面积;多西他赛使用剂量、多西他赛抽血时间点及血药浓度、化疗前患者肝功能相关指标:白蛋白、谷草转氨酶、谷丙转氨酶、总胆红素、直接胆红素;化疗前患者肾功能血清肌酐值、化疗前白细胞及中性粒细胞绝对值。采用CTCAE4.0评价标准进行不良反应评价,观察多西他赛化疗后出现的不良反应:恶心呕吐、骨髓抑制、便秘、肝功能损伤。
-
多西他赛血药浓度监测抽血时间点[8,15–19]:血样1:多西他赛滴注结束时;血样2:多西他赛滴注结束后30 min。从输液的对侧肢体通过外周静脉采集3 ml血液于EDTA紫色抽血管中。多西他赛血药浓度的监测方法由湖南德米特仪器有限公司提供,并采用湖南德米特仪器有限公司全自动二维液相色谱仪器进行分析[8,19-20]。采用Saladax公司提供的人群PK模型软件,根据多西他赛剂量、血样采集时间和多西他赛血药浓度计算AUC[16]。
-
采用风锐统计软件进行数据处理与分析[21]。正态分布计量资料以均数 ± 标准差(
$\bar x \pm \mathrm{s}$ )表示,采用t检验;计数资料以相对数构成比(%)或率(%)表示,采用χ2检验;不服从正态分布的计量资料以中位数(四分位数)[M (P25,P75)]表示,采用Mann-Whitney U检验。多西他赛AUC的影响因素采用单因素线性回归法进行分析,以P<0.05为差异具有统计学意义。 -
对2019年1月至2021年11月在我院进行了多西他赛血药浓度监测的乳腺癌患者进行筛选,24例患者纳入本研究,其中肾功能衰竭组5例,肾功能正常组19例,均为女性。患者年龄(54.7±9.3)岁,体表面积[1.6(1.5, 1.6)] m2,体重(51.5±5.1)kg,给药剂量[73.6(72.3,80.0)] mg/m2。患者体表面积、给药剂量、体重无统计学差异。两组患者在使用多西他赛化疗前肝功能相关指标及血象白细胞、中性粒细胞均在正常范围内(见表1)。
表 1 两组患者临床特征描述
临床特点 总人数(n=24) 肾功能衰竭组(n=5) 肾功能正常组(n=19) P 检验值 年龄(岁) 54.7±9.3 59.2±6.1 53.5±9.8 0.23 1.53 体表面积(m2) 1.6 (1.5, 1.6) 1.4 (1.4, 1.5)▲ 1.6 (1.5, 1.6) 0.05 3.86 给药剂量(mg/m2) 73.6(72.3,80.0) 70.4 (69.4, 73.0) 74.4(72.3, 91.2) 0.08 3.06 体重(kg) 51.5±5.1 51.4±3.8 51.5±5.5 0.97 0.001 白蛋白(g/L) 42.4±3.8 39.0±4.9▲ 43.2±3.0 0.02 6.12 谷草转氨酶(U/L) 24.6 (19.9, 31.7) 19.3 (19.1, 20.6)▲ 29.0(24.1, 34.9) 0.01 6.02 谷丙转氨酶(U/L) 21.4(15.0, 34.1) 13.7(11.2, 14.8)▲▲ 24.0(20.4, 39.4) 0.002 10.01 总胆红素(μmol/L) 11.0±3.7 9.0±2.9 11.5±3.7 0.17 1.95 直接胆红素(μmol/L) 2.4±1.1 1.9±0.8 2.5±1.1 0.27 1.28 肌酐(μmol/L) 60.5 (52.0, 285.8) 908.0 (819.0, 1 018.0)▲▲ 54.8 (52.0, 65.0) < 0.001 11.42 肌酐清除率(ml/min) 69.3(17.9, 91.8) 4.9(4.3, 5.4)▲▲ 86.3(59.3, 92.5) < 0.001 11.4 化疗前白细胞(×109/L) 6.5±1.7 7.5±1.7 6.2±1.6 0.12 2.56 化疗前中性粒绝对值(×109/L) 4.0(3.0, 4.9) 4.1(4.0, 8.7) 3.9 (2.5, 4.8) 0.21 1.55 ▲P<0.05, ▲▲P<0.01,与肾功能正常组比较。 -
表2中患者使用多西他赛的给药剂量73.6(72.3,80.0)mg/m2,两组间给药剂量无统计学意义。患者21天使用1次多西他赛,每次使用多西他赛剂量相同,因此每个患者4个多西他赛周期只需监测1次多西他赛血药浓度即可。其中两组多西他赛在2个时间点的血药浓度和多西他赛的AUC差异并无统计学意义。
表 2 24例患者使用多西他赛剂量和血药浓度及AUC情况
指标 总人数(n=24) 肾功能衰竭组(n=5) 肾功能正常组(n=19) P 检验值 多西他赛给药剂量(mg/m2) 73.6(72.3, 80.0) 70.4(69.4, 73.0) 74.4(72.3, 91.2) 0.08 3.06 多西他赛滴注结束时血药浓度(ng/ml) 682.4(460.2, 1 208.0) 650.0 (625.3, 750.0) 690.0(416.5, 1 239.0) 0.64 0.21 多西他赛滴注结束30min后血药浓度(ng/ml) 136.5±77.1 172.0±81.7 127.2±75.4 0.26 1.36 多西他赛AUC值(mg·h/L) 1.8±0.7 1.6±0.6 1.8±0.8 0.60 0.29 -
线性单因素回归分析显示,多西他赛滴注结束时血药浓度与多西他赛的AUC有显著相关性(P<0.001)。而患者年龄、给药剂量、肝功能、肾功能、化疗前白细胞及中性粒细胞绝对值均与多西他赛AUC的水平没有相关性(P>0.05)(表3)。此分析结果说明患者肾功能衰竭并不影响多西他赛AUC水平。
表 3 多西他赛 AUC 影响因素的单因素线性回归分析
因素 β 95%CI P(t检验) 年龄(岁) −0.01 −0.04~0.03 0.59 体表面积(m2) 1.09 −2.59~4.76 0.55 给药剂量(mg/m2) 0.01 −0.02~0.05 0.45 多西他赛滴注结束时血药浓度(ng/ml) 0.07▲▲ 0.04~0.09 < 0.001 多西他赛滴注结束30min后血药浓度(ng/ml) 0.34 −0.06~0.75 0.09 白蛋白(g/L) 0.05 −0.04~0.13 0.27 谷草转氨酶(U/L) −0.27 −2.72~2.17 0.82 谷丙转氨酶(U/L) −0.01 −0.02~0.01 0.46 总胆红素(μmol/L) −0.07 −0.15~0.02 0.12 直接胆红素(μmol/L) −0.23 −0.52~0.06 0.12 肌酐(μmol/L) −0.02 −0.12~0.07 0.61 体重(kg) 0.02 −0.05~0.08 0.63 肌酐清除率(ml/min) 0.14 −0.76~1.05 0.74 化疗前白细胞(×109/L) 0.02 −0.17~0.22 0.82 化疗前中性粒细胞(×109/L) 0.41 −8.34~19.15 0.97 ▲▲P<0.01,与肾功能正常组比较。 -
观察两组患者使用多西他赛后不良反应:恶心呕吐、骨髓抑制、便秘及肝功能损伤的发生率,两组间各类不良反应发生率差异无统计学意义(P>0.05),且两组患者均未出现肝功能损伤(见表4)。
表 4 两组患者使用多西他赛后不良反应发生情况
不良反应 总人群 肾功能衰竭组(n=5) 肾功能正常组(n=19) P 检验值 恶心呕吐,n (%) 0.63 Fisher I 13 (54.2) 2 (40) 11 (57.9) II 11 (45.8) 3 (60) 8 (42.1) 骨髓抑制, n (%) 0.22 Fisher I 19 (79.2) 3 (60) 16 (84.2) II 4 (16.7) 1 (20) 3 (15.8) III 1 ( 4.2) 1 (20) 0 (0) 便秘, n (%) 1 Fisher I 24 (100.0) 5 (100) 19 (100) -
多西他赛的药动学符合三房室模型[22],α半衰期为4.5 min,β半衰期为38.3 min,γ半衰期为12.2 h。它具有线性药动学特征,其峰值水平随着给药剂量和给药程序的不同而不同[23-24],AUC与给药剂量成比例增加。多西他赛主要经肝脏P450 CYP3A4系统代谢,经胆汁排泄到粪便,肾脏排泄量较小[25]。虽然多西他赛在肾脏排泄量小,但肾功能不全仍可能影响药物在体内的累积,增加药物暴露,从而导致不良反应增加。目前在进行透析的肾衰竭患者中,多西他赛说明书和指南并没有相关的剂量推荐。
本研究为单中心、回顾性病例对照研究,根据患者肾功能的状态,将患者分为肾功能衰竭组和肾功能正常组进行对比研究。可观察到:(1)两组患者化疗前临床基线特征:肝功能、血常规、体表面积及给药剂量无明显差异。(2)两组患者使用多西他赛后体内暴露量AUC并无明显差异,对多西他赛AUC影响因素进行单因素线性回归分析发现患者肾功能状态肌酐清除率并不影响其多西他赛的AUC水平。(3)对两组患者的不良反应进行随访,其血液学毒性、消化道毒性、肝毒性也无明显差异。因此本研究证实了在慢性肾功能衰竭的患者中多西他赛的暴露量及不良反应与肾功能正常的患者并无明显差异。对于慢性肾衰竭患者给予多西他赛进行化疗时,剂量可按照肾功能正常的患者进行给药。本研究对于慢性肾功能衰竭而同时患有癌症需使用化疗药物多西他赛的患者的剂量指导具有重要的临床意义。
Effect of renal failure on docetaxel exposure and adverse reactions in breast cancer patients
-
摘要:
目的 探讨肾衰竭对乳腺癌患者多西他赛的药时曲线下面积(AUC)和不良反应是否有影响,以便对此类患者的多西他赛用药提供依据。 方法 采用回顾性研究方法,选择2019年1月至2021年11月我院收治的24例已行乳腺癌根治术的使用AC-T辅助化疗的乳腺癌患者。根据肾功能病例分为两组,肾衰竭组5例、肾功能正常组19例。收集两组患者的临床特征,包括性别、年龄、体重、体表面积;多西他赛使用剂量、血药浓度、药时曲线下面积;化疗前患者肝肾功能、白细胞计数及中性粒细胞绝对值。采用单因素线性回归分析多西他赛AUC的影响因素。收集患者使用多西他赛化疗后的不良反应:恶心呕吐、骨髓抑制、便秘及肝功能损伤。以CTCAE 4.0评价标准进行不良反应评价。 结果 肾功能衰竭组与肾功能正常组的临床特征:肌酐[908.0(819.0, 1018.0)μmol/L、54.8(52.0, 65.0)μmol/L]、肌酐清除率[4.9(4.3, 5.4)ml/min、86.3(59.3, 92.5)ml/min],差异有统计学意义(P<0.001)。其他临床特征:体表面积[1.4(1.4, 1.5)m2、1. 6(1.5, 1.6)m2]、多西他赛给药剂量[70.4(69.4, 73.0)mg/m2、74.4(72.3, 91.2)mg/m2]、体重[(51.4±3.8)kg、(51.5±5.5)kg],差异均无统计学意义(P>0.05)。多西他赛化疗前肝功能及白细胞、中性粒细胞绝对值均在正常范围内。两组患者使用多西他赛后AUC值[(1.6±0.6)mg·h/L、(1.8±0.8)mg·h/L]差异无统计学意义(P>0.05)。多西他赛AUC的线性单因素回归分析显示多西他赛滴注结束时的血药浓度与其有显著相关性(P< 0.001),而其患者体表面积、多西他赛给药剂量、体重、肝肾功能均与多西他赛AUC无相关性(P>0.05)。多西他赛化疗后两组患者不良反应:恶心呕吐(I度发生率:40%、57.9%,II度发生率:60%、42.1%)、骨髓抑制(I度发生率:60%、84.2%,II度发生率:20%、15.8%)、便秘(均出现轻度便秘)的发生率差异均无统计学意义(P>0.05)。 结论 肾功能衰竭并不影响乳腺癌患者多西他赛的暴露及使用多西他赛后出现的不良反应。 Abstract:Objective To investigate the influence of renal failure on the area under curve (AUC) and adverse reactions of docetaxel in breast cancer patients, and provide evidence for the dosage of docetaxel in renal failure patients. Methods A retrospective study was conducted on 24 patients with breast cancer who had undergone radical mastectomy and received AC-T adjuvant chemotherapy in our hospital from January 2019 to November 2021. According to renal function cases, the patients were divided into two groups: renal failure group (n=5) and normal renal function group (n=19). The clinical characteristics such as gender, age, body weight and body surface area of patients in two groups, docetaxel dose, blood concentration, area under the curve, liver and kidney function, white blood cell count and absolute value of neutrophil before chemotherapy were collected. Single factor linear regression was used to analyze the influencing factors of the AUC of docetaxel. Adverse reactions after chemotherapy with docetaxel including nausea and vomiting, bone marrow suppression, constipation and liver function injury were collected. CTCAE 4.0 evaluation standard was used to evaluate adverse reactions. Results The clinical characteristics of creatinine [908.0 (819.0, 1018.0) μmol/L vs 54.8 (52.0, 65.0) μmol/L] and creatinine clearance rate [4.9 (4.3, 5.4) ml /min vs 86.3 (59.3, 92.5) ml/min] of the renal failure group and the normal renal function group have significant difference (P<0.001), while no significant difference (P>0.05) were found in the body surface area [1.4 (1.4, 1.5) m2 vs 1. 6 (1.5, 1.6) m2], docetaxel dose [70.4 (69.4, 73.0) mg/m2 vs 74.4 (72.3, 91.2) mg/m2], body weight [(51.4±3.8) kg vs (51.5±5.5) kg]. Liver function, white blood cells and neutrophils were within the normal range before chemotherapy with docetaxel. There was no significant difference in AUC value [(1.6±0.6) mg·h/L vs (1.8±0.8) mg·h/L] between the two groups after chemotherapy with docetaxel (P>0.05). Linear univariate regression analysis indicated that the blood concentration at the end of docetaxel infusion was significantly associated with AUC of docetaxel (P<0.001), while the body surface area, dose of docetaxel, body weight, liver and kidney function were not correlated with AUC of docetaxel (P>0.05). After chemotherapy with docetaxel, adverse reactions of patients in the two groups: nausea and vomiting (grade I incidence: 40% vs. 57.9%, grade II incidence: 60% vs. 42.1%), myelosuppression (grade I incidence: 60% vs. 84.2%, grade II incidence: 20% vs 15.8%) and constipation (all mild constipation) had no significant difference (P>0.05). Conclusion Renal failure did not affect the exposure of docetaxel and the adverse reactions after chemotherapy with docetaxel in breast cancer patients. -
Key words:
- renal failure /
- breast cancer /
- docetaxel /
- AUC /
- adverse reaction
-
肺型氧中毒是长期吸入高浓度氧气导致的肺部损伤,表现为肺组织充血、水肿、炎性浸润等,其中以多型核白细胞为主[1]。活化的多型核白细胞可以激活氧化酶,进而产生和释放大量的炎症介质和氧自由基,进一步介导级联式扩大的炎症反应对肺部造成损伤[2]。在饱和潜水或重型减压病患者进行加压治疗时,肺型氧中毒已成为最常见的并发症[3]。然而,目前关于高压氧(HBO)诱导的肺型氧中毒发生机制缺乏深入的探讨。
山莨菪碱(ANI)是一种毒蕈碱(M)受体阻滞剂,新斯的明(NEO)是一种临床常见的胆碱酯酶抑制剂。我们课题组前期研究发现ANI和NEO以500∶1的比例组成复方,简称新斯莨菪碱,能协同地激活α7nAChR,并减轻二者联用所致的不良反应。研究表明激活α7nAChR受体在感染性休克[4]、类风湿关节炎[5]、挤压综合征[6]、脑卒中[7]等动物模型上均发挥保护作用。然而,在HBO诱导的肺型氧中毒的发生、发展过程中,新斯莨菪碱是否可以用于肺型氧中毒的治疗?其治疗作用的病理生理机制是什么?这些都尚未阐明。
铁死亡是一种以细胞内铁依懒性的活性氧(ROS)异常增高,导致细胞内ROS的生成和降解失衡为特征的细胞死亡方式[8]。高浓度氧暴露条件下生成的大量ROS,一方面可直接对肺细胞产生损害,另一方面可参与炎症反应,促进炎症细胞渗透聚集,使各种致炎因子的表达增多,进一步加重肺的急性炎症反应[9]。
本文探究了新斯莨菪碱对HBO诱导的肺型氧中毒是否具有保护效应,并在此基础上,阐明铁死亡在新斯莨菪碱治疗HBO诱导的肺型氧中毒中的作用,以期为肺型氧中毒的临床防治提供新的理论指导和思路。
1. 材料和方法
1.1 实验动物
雄性C57BL/6小鼠,6~8周龄,小鼠购自上海斯莱克公司。所有小鼠均饲养于22℃,昼夜循环12 h的独立饲养系统中。小鼠自由饮食、自由饮水。所有实验均经海军军医大学实验动物伦理委员会批准并按照指南进行。
1.2 仪器和试剂
小型实验动物氧舱(RDC150-300-6,海军军医大学);台式高速冷冻离心(SCILOGEX);7500RT-PCR 仪器(Applied Biosystems 公司);激光共聚焦显微镜(日本Olympus公司);酶标仪(瑞士Tacan 公司);高速组织研磨仪(上海净信实业发展有限公司);Trizol、BCA 法蛋白定量试剂盒(美国GLPBIO公司);苏木精-伊红(上海Sangon Biotech公司);戊巴比妥(上海国药集团化学试剂公司);RT-PCR试剂盒(日本Takara公司);组织铁检测试剂盒(北京Solarbiog公司);伊文思蓝(美国Sigma-Aldrich公司);MDA、GSH检测试剂盒(上海Beyotime公司);Perls stain、抗谷胱甘肽过氧化物酶4(GPX4)抗体、抗β-actin抗体(英国Abcam公司);驴抗兔/驴抗鼠二抗、Odyssey 扫膜仪(LI-COR公司)。
1.3 实验方法
1.3.1 肺型氧中毒模型的制备
将小鼠置于动物加压舱内,先用纯氧冲洗舱5 min,然后将压力在5 min内调至舱内为100% O2、2.5 ATA,维持6 h,在暴露过程中保持 0.5 L/min的连续氧流量,每1~2 h调整气阀进行快速通气,同时舱内放置碱石灰以避免呼吸产生的CO2在舱内蓄积。动物实验分组如下:①正常组:腹腔注射给予生理盐水10 ml/kg,常压饲养;②模型组:HBO暴露前30 min给予生理盐水10 ml/kg,2.5 ATA暴露6 h;③新斯莨菪碱组:HBO暴露前30 min给予ANI(25 mg/kg)+NEO(50 μg/kg),2.5 ATA 暴露6 h。
1.3.2 HE染色
使用戊巴比妥钠麻醉并处死小鼠,解剖取小鼠左肺组织于4%多聚甲醛中固定,然后用石蜡包埋并切成5 μm的薄片。在 200倍光学显微镜下观察组织炎症、水肿或其他损伤。组织学评分:0分为正常组织学;1分代表轻度白细胞浸润和毛细血管充血;2分代表轻度白细胞浸润、血管周围水肿、肺结构部分破坏和出血;3分代表强烈的白细胞浸润和肺结构破坏。
1.3.3 伊文思蓝染色
HBO暴露6 h后,将小鼠固定,尾静脉注射1%伊文思蓝(50 mg/kg)后放回笼中自由活动,约30 min处死动物,用PBS灌流后取肺组织,用滤纸擦干肺组织表面水分并对肺组织进行拍照。每100 mg肺组织加入2 ml甲酰胺,55℃孵育18 h,反应结束后10 000×g 离心30 min后取上清液,并在630、740 nm处测量吸光度。
1.3.4 肺组织湿干比
将小鼠麻醉处死后,取新鲜左肺组织,滤纸擦干表面血迹后称重记为湿重,随后将肺组织置于60℃干燥箱中,每日测量肺组织重量,直至肺组织重量不再变化,此时肺组织重量为干重。肺含水量(%)=(湿重−干重)/湿重×100%。
1.3.5 肺泡灌洗液蛋白含量测定
用1 ml预冷PBS经气管插管灌洗肺3次,收集混合的支气管肺泡灌洗液(BALF),将收集到的BALF以1 500 r/min 离心10 min获得其上清液,BCA法测定试剂盒定量BALF中总蛋白。
1.3.6 非血红素铁含量测定
30 mg肺组织在冷PBS中清洗,在冰上用匀浆器在铁测定缓冲液中匀浆,在4℃以16 000×g离心10 min,收集上清液用铁检测试剂盒进行组织铁含量测定。
1.3.7 普鲁士蓝染色
戊巴比妥钠麻醉处死小鼠,取新鲜肺组织于4%多聚甲醛中固定,固定48 h后进行梯度脱水,二甲苯置换,浸蜡;包埋、切片(厚度为5 μm)。将石蜡切片脱蜡后用Prussian Blue Stain(等体积的亚铁氰化钾溶液和盐酸溶液混合,制成铁染色工作液)染色15 min,洗涤,然后用核固红溶液染色5 min,洗涤,封片后在Olympus光学显微镜下观察染色切片。
1.3.8 丙二醛(MDA)的测定
取100 μg肺组织于300 μl PBS溶液中,匀浆机研磨后,以12 000 r/min离心10 min后取上清液待测。所有试剂盒均按照制造商的说明书使用,按检测工作液与上清2∶1配好检测体系后100℃水浴15 min,冷却后于532 nm处测吸光度。
1.3.9 谷胱甘肽(GSH)的测定
取100 μg肺组织,与700 μl蛋白去除试剂S溶液混匀,匀浆机研磨后以12 000 r/min,离心10 min后取上清液进行总谷胱甘肽测定。所有试剂盒均按照制造商的说明书使用,25℃反应60 min即可测得氧化性谷胱甘肽(GSSG)含量,还原GSH含量=总谷胱甘肽含量−GSSG×2 。
1.3.10 实时定量PCR
动物出舱后,解剖取肺组织,使用TRIzol试剂提取组织总RNA并逆转录成cDNA,进行PCR扩增,采用2–ΔΔCT分析目的基因的相对表达量,引物序列见表1。
表 1 实时定量PCR引物序列引物名称 引物序列(5′—3′) IL-6(F) CCAGAAACCGCTATGAAGTTCC IL-6(R) GTTGGGAGTGGTATCCTCTGTGA IL-1β(F) GTTCCCATTAGACAACTGCACTACAG IL-1β(R) GTCGTTGCTTGGTTCTCCTTGTA TNF-α(F) CCCCAAAGGGATGAGAAGTTC TNF-α(R) CCTCCACTTGGTGGTTTGCT GAPDH(F) GTATGACTCCACTCACGGCAAA GAPDH(R) GGTCTCGCTCCTGGAAGATG 注:F: 正向引物; R: 反向引物。 1.3.11 Western blot
提取肺组织蛋白,BCA法测定蛋白浓度。十二烷基硫酸钠-聚丙烯酰胺凝胶(SDS-PAGE)电泳分离蛋白质,然后转移到硝酸纤维素膜上,与GPX4、β-肌动蛋白共孵育。最后,根据情况将膜与驴抗兔或驴抗鼠二抗孵育,使用Odyssey 扫膜仪获取图像,通过Image J 软件对条带进行定量分析。
1.4 统计学处理
实验数据的分析以及数据处理均使用GraphPad Prism 8.0 软件进行。所有值以(
$ \bar{x}\pm s $ )表示,多组间比较用单因素方差分析(ANOVA)并 Bonferroni 检验,P<0.05 视为差异具有统计学意义。2. 结果
2.1 新斯莨菪碱保护了 HBO暴露引起的肺组织损伤
首先,探究HBO暴露后对肺组织的病理损伤情况以及新斯莨菪碱对HBO诱导的肺损伤的作用。将C57BL/6小鼠置于2.5 ATA的高压氧舱中暴露6 h建立肺型氧中毒模型,然后通过HE染色观察肺组织结构变化,如图1所示。正常组小鼠肺泡肺泡壁结构完整,间隔无增厚,肺泡腔内无渗出物;HBO暴露后,肺泡间隔出现水肿、增厚,肺泡腔内可见红细胞渗出,肺间质见大量炎性细胞浸润(图1B);给予新斯莨菪碱治疗后,肺水肿明显减轻,肺泡内炎性细胞浸润以及红细胞渗出减少,肺组织病理损伤减轻(图1C~D)。以上结果表明新斯莨菪碱治疗能减轻肺型氧中毒后肺组织的病理损伤。
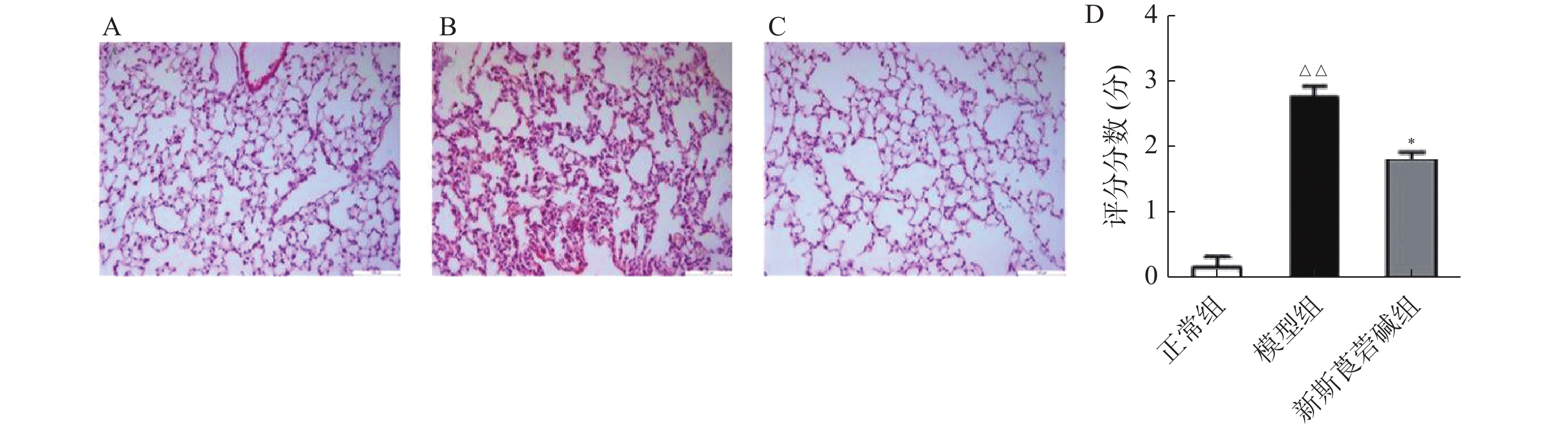 图 1 新斯莨菪碱对高压氧诱导的肺损伤的作用(n=3,重复3次)A−C. 小鼠肺组织HE染色;D. 肺组织病理学损伤评分*P<0.05,与模型组比较;△△P<0.01,与正常组比较。
图 1 新斯莨菪碱对高压氧诱导的肺损伤的作用(n=3,重复3次)A−C. 小鼠肺组织HE染色;D. 肺组织病理学损伤评分*P<0.05,与模型组比较;△△P<0.01,与正常组比较。2.2 新斯莨菪碱改善肺组织渗透性
从图2可以看出,模型组伊文思蓝染色加深,表明HBO暴露后肺渗透性增加,而新斯莨菪碱治疗后上述情况明显改善(图2A~B)。与正常组相比,模型组肺组织湿干比重明显增加,而新斯莨菪碱治疗后能够显著降低肺湿干比(图2C)。此外,HBO暴露后肺泡灌洗液中蛋白含量升高,新斯莨菪碱治疗能够有效减少肺泡灌洗液中蛋白的渗出(图2D)。这些结果表明,新斯莨菪碱治疗能够有效改善肺型氧中毒后的肺水肿及肺组织渗出。
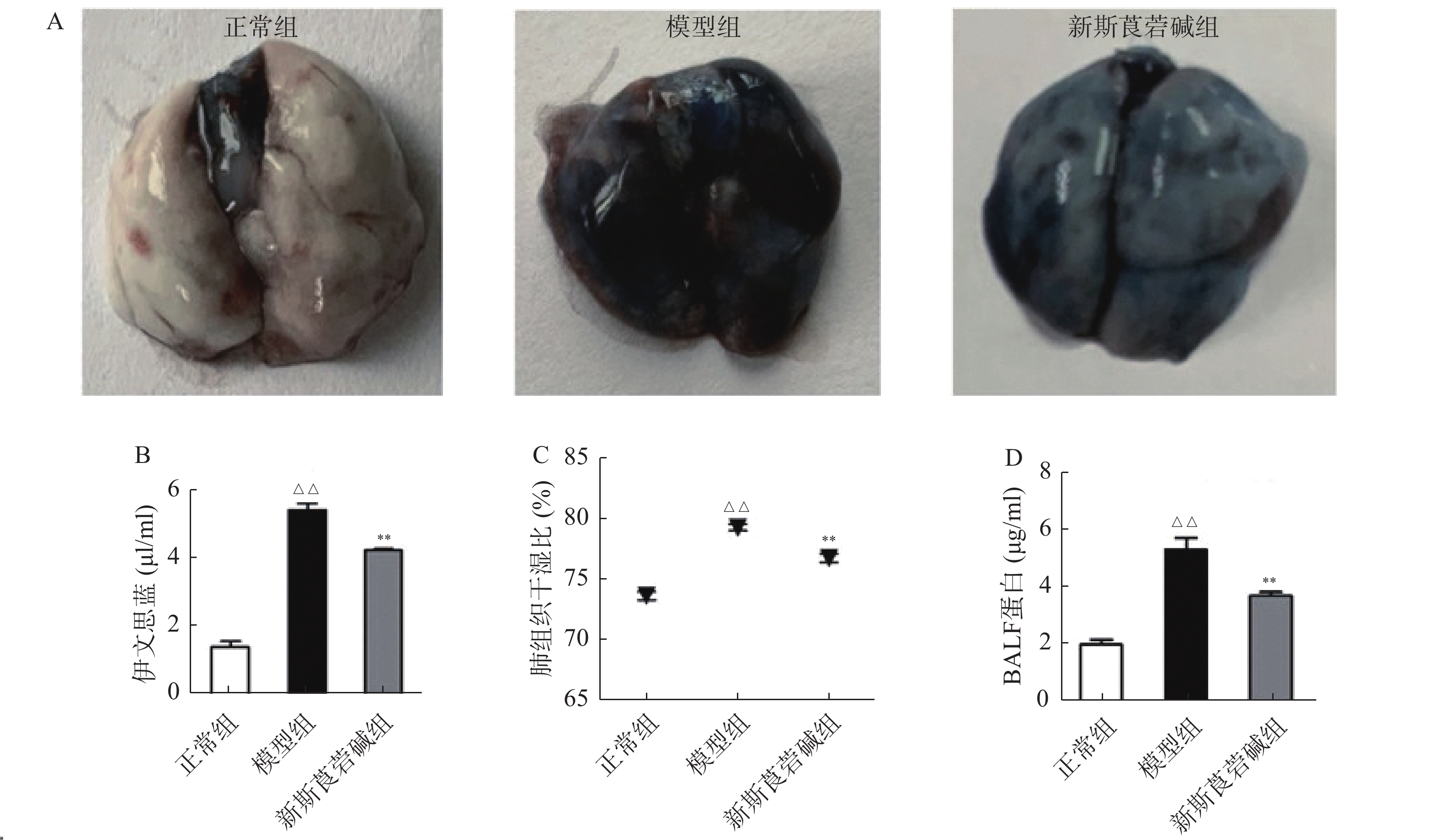 图 2 新斯莨菪碱对肺组织渗透性的影响(n=3,重复3次)A. 不同组别肺组织伊文思蓝染色后;B. 肺组织伊文思蓝定量检测;C. 肺组织干湿比;D. 肺泡灌洗液蛋白含量测定**P<0.01,与模型组比较;△△P<0.01,与正常组比较。
图 2 新斯莨菪碱对肺组织渗透性的影响(n=3,重复3次)A. 不同组别肺组织伊文思蓝染色后;B. 肺组织伊文思蓝定量检测;C. 肺组织干湿比;D. 肺泡灌洗液蛋白含量测定**P<0.01,与模型组比较;△△P<0.01,与正常组比较。2.3 新斯莨菪碱抗炎、抗氧化应激作用
从图3中可以看出,HBO暴露后,肺组织中促细胞因子,如TNF-α、IL-1β、IL-6和IFN-γ的mRNA显著增高,抗炎因子IL-4及TGF-β则显著降低,而新斯莨菪碱能显著降低促炎因子的表达,同时提高抗炎因子的水平(图3A~F)。HBO暴露后肺组织氧化指标MDA升高,而抗氧化指标GSH降低,给予新斯莨菪碱治疗后,能明显逆转上述变化(图3G~H)。由此说明,新斯莨菪碱能降低HBO诱导的肺型氧中毒引起的炎症反应及氧化应激水平。
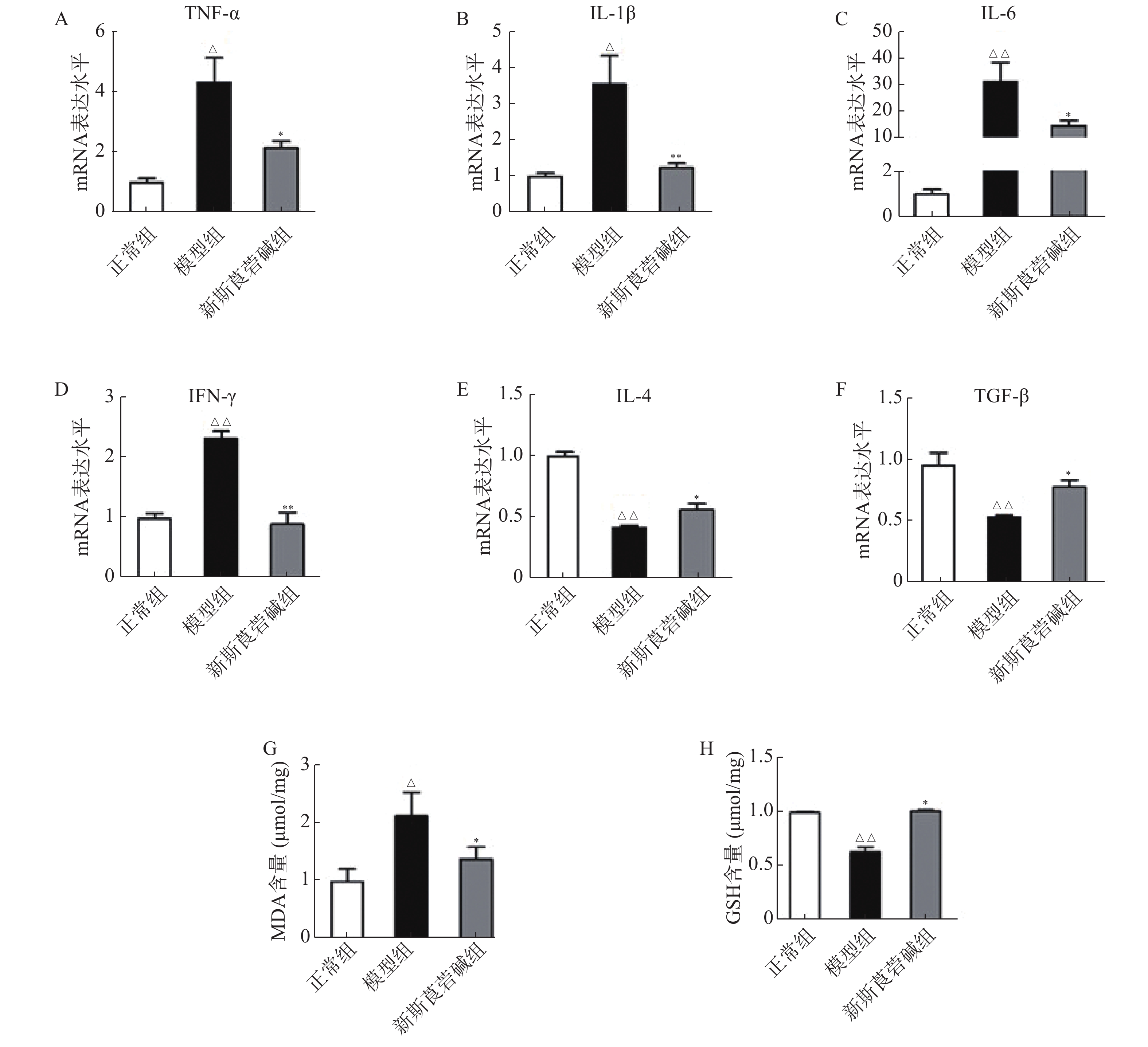 图 3 新斯莨菪碱治疗对炎症因子mRNA水平的影响(n=3,重复3次)A−F. 肺组织中TNF-α、IL-1β、IL-6、IFN-γ、IL-4和TGF-β的mRNA水平的表达;G. MDA含量;H. GSH含量*P<0.05,**P<0.01,与模型组比较;△P<0.05,△△P<0.01,与正常组比较。
图 3 新斯莨菪碱治疗对炎症因子mRNA水平的影响(n=3,重复3次)A−F. 肺组织中TNF-α、IL-1β、IL-6、IFN-γ、IL-4和TGF-β的mRNA水平的表达;G. MDA含量;H. GSH含量*P<0.05,**P<0.01,与模型组比较;△P<0.05,△△P<0.01,与正常组比较。2.4 新斯莨菪碱减轻铁死亡
铁元素是维持正常生理活动所必需的,但过量的铁会产生对细胞有害的自由基。在病理状态下,铁蛋白的破坏导致细胞内游离铁的增高,脂质过氧化反应和游离铁的增高会导致细胞的铁死亡。如图4所示,HBO暴露后普鲁士蓝染色可见肺组织中铁含量增高,对肺组织铁离子含量进行检测得到了同样的结果(图4A~C)。GPX4是一种重要的抗氧化酶,HBO暴露后GPX4的表达降低(图4D~E),导致机体抗氧化应激能力减弱,对细胞有害的自由基无法被有效清除。而新斯莨菪碱治疗后可以改善GPX4的表达,降低肺组织铁含量,从而减少细胞铁死亡的发生,对肺组织起到保护作用。
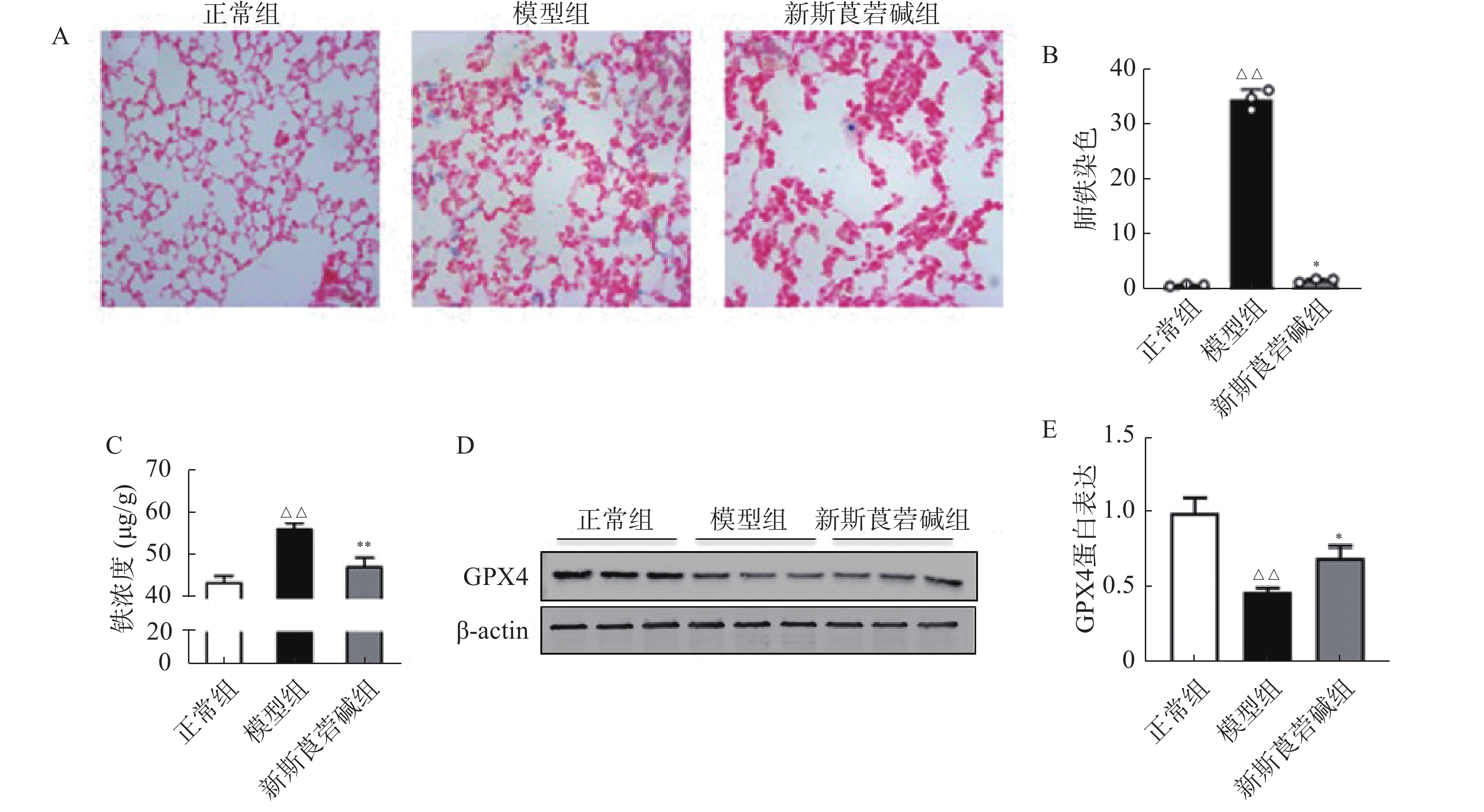 图 4 铁死亡途径在新斯莨菪碱治疗HBO诱导的肺型氧中毒的作用(n=3,重复3次)A. 不同组别肺组织普鲁士蓝染色;B. 肺铁染色;C. 肺组织铁含量;D. 肺组织GPX4的表达*P<0.05,**P<0.01,与模型组比较;△P<0.05,△△P<0.01,与正常组比较。
图 4 铁死亡途径在新斯莨菪碱治疗HBO诱导的肺型氧中毒的作用(n=3,重复3次)A. 不同组别肺组织普鲁士蓝染色;B. 肺铁染色;C. 肺组织铁含量;D. 肺组织GPX4的表达*P<0.05,**P<0.01,与模型组比较;△P<0.05,△△P<0.01,与正常组比较。3. 讨论
HBO已被广泛用于多种疾病的治疗以及潜水作业中,并发挥了难以替代的作用[10]。然而,在饱和潜水或重型减压病患者加压治疗以及临床上用于治疗新生儿呼吸窘迫时,HBO诱导的肺损伤已成为最常见的并发症[11]。研究发现,当HBO环境持续存在时,将出现小气道功能障碍,表现为肺活量降低、气体扩散功能减弱和肺顺应性降低[12]。与之前的报道一致,本研究结果表明,与正常组相比,模型组的肺组织损伤显著加重;伊文思蓝染色显示HBO暴露后染色加深,肺湿/干比增加,同时肺泡灌洗液中的蛋白含量也显著增加,新斯莨菪碱治疗后这些变化被显著抑制,这些结果初步表明新斯莨菪碱与HBO诱导的肺型氧中毒的发生密切相关。
ANI属于传出自主神经M受体阻断剂,阻断M受体引起交感神经优势。NEO是一种可逆性胆碱酯酶抑制剂,可有效延长乙酰胆碱的作用时间,同时激活胆碱能抗炎通路[11]。Sun等[4]研究证实,两药物联合使用,不仅可以加强胆碱能抗炎通路的炎症抑制作用,还可降低NEO的不良反应,达到协同增强作用。本课题组前期的研究发现ANI和NEO联合使用,能够通过阻断巨噬细胞上的毒蕈碱型受体,使更多的乙酰胆碱作用于α7尼古丁乙酰胆碱受体,进而对胆碱能抗炎通路起到双向活化调节作用,进一步加强了对炎症反应的抑制作用[7]。本研究发现新斯莨菪碱能够显著降低肺组织中促炎因子TNF-α、IL-1β、IL-6和IFN-γ的表达。由此说明,新斯莨菪碱可减轻HBO导致的肺部炎症反应。
肺型氧中毒包括高压力损伤和高浓度氧损伤,目前认为ROS 及其代谢产物介导的生化紊乱是导致肺型氧中毒发生发展的主要因素。HBO环境下会产生大量氧自由基,可能导致细胞因子释放和炎症反应的增加,而异常的炎症反应可加剧肺损伤[13]。研究发现,HBO 暴露后肺组织中氧化指标MDA 显著升高、抗氧化指标 GSH 则显著降低,而给新斯莨菪碱治疗能明显改善肺组织氧化损伤情况。
铁死亡作为一种新发现的细胞死亡方式,其释放出内源性损伤相关分子(DAMPs)与炎症和氧化应激之间存在着诸多途径的交互作用。GPX4作为铁死亡的核心调控因子,近年来受到广泛关注。研究表明GPX4的缺乏或功能丧失会加剧铁死亡的发生,并促使细胞进一步受到氧化应激的影响[14]。Kang等[15]的研究发现在LPS诱导的脓毒症模型中,敲除GPX4导致脂质过氧化的加剧并进一步促进铁死亡的发生。本研究发现,HBO暴露后肺组织中铁含量增高、GPX4的表达量降低,而新斯莨菪碱治疗后可以逆转这些变化。
综上所述,新斯莨菪碱可能通过激活胆碱能抗炎通路,从而抑制炎症的和氧化应激的发生,进而减少了肺组织中游离铁的含量,最终抑制细胞铁死亡。下一步我们将通过体外实验,进一步验证在HBO诱导的肺型氧中毒模型中,新斯莨菪碱是否通过抑制铁死亡的产生从而减轻肺组织的损伤情况,以期为防治HBO诱导的肺型氧中毒提供新的策略。
-
表 1 两组患者临床特征描述
临床特点 总人数(n=24) 肾功能衰竭组(n=5) 肾功能正常组(n=19) P 检验值 年龄(岁) 54.7±9.3 59.2±6.1 53.5±9.8 0.23 1.53 体表面积(m2) 1.6 (1.5, 1.6) 1.4 (1.4, 1.5)▲ 1.6 (1.5, 1.6) 0.05 3.86 给药剂量(mg/m2) 73.6(72.3,80.0) 70.4 (69.4, 73.0) 74.4(72.3, 91.2) 0.08 3.06 体重(kg) 51.5±5.1 51.4±3.8 51.5±5.5 0.97 0.001 白蛋白(g/L) 42.4±3.8 39.0±4.9▲ 43.2±3.0 0.02 6.12 谷草转氨酶(U/L) 24.6 (19.9, 31.7) 19.3 (19.1, 20.6)▲ 29.0(24.1, 34.9) 0.01 6.02 谷丙转氨酶(U/L) 21.4(15.0, 34.1) 13.7(11.2, 14.8)▲▲ 24.0(20.4, 39.4) 0.002 10.01 总胆红素(μmol/L) 11.0±3.7 9.0±2.9 11.5±3.7 0.17 1.95 直接胆红素(μmol/L) 2.4±1.1 1.9±0.8 2.5±1.1 0.27 1.28 肌酐(μmol/L) 60.5 (52.0, 285.8) 908.0 (819.0, 1 018.0)▲▲ 54.8 (52.0, 65.0) < 0.001 11.42 肌酐清除率(ml/min) 69.3(17.9, 91.8) 4.9(4.3, 5.4)▲▲ 86.3(59.3, 92.5) < 0.001 11.4 化疗前白细胞(×109/L) 6.5±1.7 7.5±1.7 6.2±1.6 0.12 2.56 化疗前中性粒绝对值(×109/L) 4.0(3.0, 4.9) 4.1(4.0, 8.7) 3.9 (2.5, 4.8) 0.21 1.55 ▲P<0.05, ▲▲P<0.01,与肾功能正常组比较。  下载: 导出CSV
下载: 导出CSV
表 2 24例患者使用多西他赛剂量和血药浓度及AUC情况
指标 总人数(n=24) 肾功能衰竭组(n=5) 肾功能正常组(n=19) P 检验值 多西他赛给药剂量(mg/m2) 73.6(72.3, 80.0) 70.4(69.4, 73.0) 74.4(72.3, 91.2) 0.08 3.06 多西他赛滴注结束时血药浓度(ng/ml) 682.4(460.2, 1 208.0) 650.0 (625.3, 750.0) 690.0(416.5, 1 239.0) 0.64 0.21 多西他赛滴注结束30min后血药浓度(ng/ml) 136.5±77.1 172.0±81.7 127.2±75.4 0.26 1.36 多西他赛AUC值(mg·h/L) 1.8±0.7 1.6±0.6 1.8±0.8 0.60 0.29
下载: 导出CSV
表 3 多西他赛 AUC 影响因素的单因素线性回归分析
因素 β 95%CI P(t检验) 年龄(岁) −0.01 −0.04~0.03 0.59 体表面积(m2) 1.09 −2.59~4.76 0.55 给药剂量(mg/m2) 0.01 −0.02~0.05 0.45 多西他赛滴注结束时血药浓度(ng/ml) 0.07▲▲ 0.04~0.09 < 0.001 多西他赛滴注结束30min后血药浓度(ng/ml) 0.34 −0.06~0.75 0.09 白蛋白(g/L) 0.05 −0.04~0.13 0.27 谷草转氨酶(U/L) −0.27 −2.72~2.17 0.82 谷丙转氨酶(U/L) −0.01 −0.02~0.01 0.46 总胆红素(μmol/L) −0.07 −0.15~0.02 0.12 直接胆红素(μmol/L) −0.23 −0.52~0.06 0.12 肌酐(μmol/L) −0.02 −0.12~0.07 0.61 体重(kg) 0.02 −0.05~0.08 0.63 肌酐清除率(ml/min) 0.14 −0.76~1.05 0.74 化疗前白细胞(×109/L) 0.02 −0.17~0.22 0.82 化疗前中性粒细胞(×109/L) 0.41 −8.34~19.15 0.97 ▲▲P<0.01,与肾功能正常组比较。
下载: 导出CSV
表 4 两组患者使用多西他赛后不良反应发生情况
不良反应 总人群 肾功能衰竭组(n=5) 肾功能正常组(n=19) P 检验值 恶心呕吐,n (%) 0.63 Fisher I 13 (54.2) 2 (40) 11 (57.9) II 11 (45.8) 3 (60) 8 (42.1) 骨髓抑制, n (%) 0.22 Fisher I 19 (79.2) 3 (60) 16 (84.2) II 4 (16.7) 1 (20) 3 (15.8) III 1 ( 4.2) 1 (20) 0 (0) 便秘, n (%) 1 Fisher I 24 (100.0) 5 (100) 19 (100)
下载: 导出CSV
-
[1] JANUS N, THARIAT J, BOULANGER H, et al. Proposal for dosage adjustment and timing of chemotherapy in hemodialyzed patients[J]. Ann Oncol,2010,21(7):1395-1403. doi: 10.1093/annonc/mdp598 [2] HERBST R S, KHURI F R. Mode of action of docetaxel - a basis for combination with novel anticancer agents[J]. Cancer Treat Rev,2003,29(5):407-415. doi: 10.1016/S0305-7372(03)00097-5 [3] 江泽飞, 李健斌. 乳腺癌诊疗指南和临床实践历程[J]. 中华外科杂志, 2020, 58(2):85-90. doi: 10.3760/cma.j.issn.0529-5815.2020.02.002 [4] CHEW S C, SINGH O, CHEN X G, et al. The effects of CYP3A4, CYP3A5, ABCB1, ABCC2, ABCG2 and SLCO1B3 single nucleotide polymorphisms on the pharmacokinetics and pharmacodynamics of docetaxel in nasopharyngeal carcinoma patients[J]. Cancer Chemother Pharmacol,2011,67(6):1471-1478. doi: 10.1007/s00280-011-1625-9 [5] ASHBEE H R, BARNES R A, JOHNSON E M, et al. Therapeutic drug monitoring (TDM) of antifungal agents: guidelines from the British Society for Medical Mycology[J]. J Antimicrob Chemother,2014,69(5):1162-1176. doi: 10.1093/jac/dkt508 [6] ALEXANDRE J, REY E, GIRRE V, et al. Relationship between cytochrome 3A activity, inflammatory status and the risk of docetaxel-induced febrile neutropenia: a prospective study[J]. Ann Oncol,2007,18(1):168-172. doi: 10.1093/annonc/mdl321 [7] BAKER SD, LI J, TEN TIJE AJ, et al. Relationship of systemic exposure to unbound docetaxel and neutropenia[J]. Clin Pharmacol Ther,2005,77(1):43-53. doi: 10.1016/j.clpt.2004.09.005 [8] SUN N, SHEN B, ZHU J L, et al. Clinical application of the AUC-guided dosage adjustment of docetaxel-based chemotherapy for patients with solid tumours: a single centre, prospective and randomised control study[J]. J Transl Med,2020,18(1):226. doi: 10.1186/s12967-020-02394-w [9] JANUS N, LAUNAY-VACHER V, DERAY G, et al. Gestion des chimiothérapies chez les patients hémodialysés[J]. Bull Cancer,2012,99(3):371-380. doi: 10.1684/bdc.2011.1483 [10] GARLAND L L, HIDALGO M, MENDELSON D S, et al. A phase I clinical and pharmacokinetic study of oral CI-1033 in combination with docetaxel in patients with advanced solid tumors[J]. Clin Cancer Res, 2006, 12(14 Pt 1): 4274-4282. [11] KUO J C, CRAFT P S. Administration of chemotherapy in patients on dialysis[J]. Anticancer Drugs,2015,26(7):779-784. doi: 10.1097/CAD.0000000000000243 [12] LABAKI C, RAWADI E, CHEBEL R, et al. Anti-neoplastic agents for patients on peritoneal dialysis: a systematic review[J]. Crit Rev Oncol Hematol,2020,150:102947. doi: 10.1016/j.critrevonc.2020.102947 [13] JANUS N, LAUNAY-VACHER V. Anticancer drugs in end-stage kidney disease patients[J]. Semin Dial,2015,28(4):413-416. doi: 10.1111/sdi.12371 [14] 中国抗癌协会乳腺癌专业委员会. 中国抗癌协会乳腺癌诊治指南与规范(2019年版)[J]. 中国癌症杂志, 2019, 29(8):609-680. doi: 10.19401/j.cnki.1007-3639.2019.08.009 [15] BAILLE P, BRUNO R, SCHELLENS J H, et al. Optimal sampling strategies for Bayesian estimation of docetaxel (Taxotere) clearance[J]. Clin Cancer Res,1997,3(9):1535-1538. [16] ENGELS F K, LOOS W J, VAN DER BOL J M, et al. Therapeutic drug monitoring for the individualization of docetaxel dosing: a randomized pharmacokinetic study[J]. Clin Cancer Res,2011,17(2):353-362. doi: 10.1158/1078-0432.CCR-10-1636 [17] ANDRIGUETTI N B, RAYMUNDO S, ANTUNES M V, et al. Pharmacogenetic and pharmacokinetic dose individualization of the taxane chemotherapeutic drugs paclitaxel and docetaxel[J]. Curr Med Chem,2017,24(33):3559-3582. [18] MA Y X, LIN Q G, YANG Y P, et al. Clinical pharmacokinetics and drug exposure-toxicity correlation study of docetaxel based chemotherapy in Chinese head and neck cancer patients[J]. Ann Transl Med,2020,8(5):236. doi: 10.21037/atm.2020.01.76 [19] 蒋侃, 黄诚, 林根, 等. 检测多西他赛血药浓度指导晚期非小细胞肺癌患者的个体化治疗[J]. 临床肿瘤学杂志, 2018, 23(7):610-614. doi: 10.3969/j.issn.1009-0460.2018.07.007 [20] RIGO-BONNIN R, COBO-SACRISTÁN S, GONZALO-DIEGO N, et al. Measurement of total and free docetaxel concentration in human plasma by ultra-performance liquid chromatography-tandem mass spectrometry[J]. J Pharm Biomed Anal,2016,117:140-149. doi: 10.1016/j.jpba.2015.08.025 [21] YANG Q L, ZHENG J Z, CHEN W Y, et al. Association between preadmission metformin use and outcomes in intensive care unit patients with Sepsis and type 2 diabetes: a cohort study[J]. Front Med (Lausanne),2021,8:640785. [22] KENMOTSU H, TANIGAWARA Y. Pharmacokinetics, dynamics and toxicity of docetaxel: why the Japanese dose differs from the Western dose[J]. Cancer Sci,2015,106(5):497-504. doi: 10.1111/cas.12647 [23] JOERGER M. Treatment regimens of classical and newer taxanes[J]. Cancer Chemother Pharmacol,2016,77(2):221-233. doi: 10.1007/s00280-015-2893-6 [24] ALKEN S, KELLY C M. Benefit risk assessment and update on the use of docetaxel in the management of breast cancer[J]. Cancer Manag Res,2013,5:357-365. [25] CLARKE S J, RIVORY L P. Clinical pharmacokinetics of docetaxel[J]. Clin Pharmacokinet,1999,36(2):99-114. doi: 10.2165/00003088-199936020-00002 -


