

 下载:
下载:

 下载:
下载:

-
肝纤维化是由肝炎病毒、酒精摄入过量或代谢紊乱引起的急性/慢性肝损伤的一种病理伤口愈合反应,也是慢性肝病发病率和病死率高的主要原因[1, 2]。肝纤维化的特点是I 型胶原和纤维连接蛋白等细胞外基质(ECM)成分的过多聚集,形成纤维疤痕扭曲肝脏结构,最终造成肝脏器官功能损伤[2]。研究显示,肝星状细胞(HSCs)的过度激活是肝纤维化进程中的关键环节,也是肝纤维化防治研究的重要靶点[3, 4]。
全反式维甲酸(ATRA)是维生素A主要的生物活性形式,已是急性早幼粒细胞白血病的标准治疗方案[5]。近期研究证实,ATRA可逆转HSCs的活化,并对肝纤维化具有抑制作用,但其具体机制尚未完全阐明[6]。本文拟在细胞水平探索ATRA抑制HSCs增殖及活化的作用和机制,为ATRA的临床应用提供理论和实验基础。
-
HSCs系LX-2细胞和培养基购自上海中乔新舟生物科技公司。胎牛血清(FBS)购自美国Gibco公司。血小板源性生长因子(PDGF-bb)、ATRA购自美国MedChemExpress公司。RNAiso试剂,逆转录和定量PCR试剂盒购自大连宝生物公司。PCR引物由上海生工生物工程公司合成。CCK-8、活性氧(ROS)、还原型谷胱甘肽(GSH)和丙二醛(MDA)等检测试剂盒购自上海碧云天生物科技公司。抗α-SMA、Collagen I、NRF2和LC3的抗体购自武汉三鹰生物科技公司;抗HO-1、ATF4、Beclin 1和GAPDH的抗兔购自武汉博士德生物科技公司。辣根过氧化物酶及FITC标记的二抗购自美国thermo Fisher Scientific公司。
-
HSCs系LX-2细胞解冻后,在DMEM培养基(含2%FBS),37 ℃,5%CO2条件下生长,每2 d更换一次培养基,细胞融合度达到85%以上时传代培养。以含PDGF-bb(10 ng/ml)的DMEM培养基作为诱导培养基;ATRA以5 μmol/L的浓度刺激。
-
细胞生长活力通过CCK-8法检测。以每孔2×103个细胞的密度将LX-2细胞接种于96孔培养板,培养过夜。分别在常规培养基(含10%FBS)和PDGF-bb诱导培养基中培养,并添加ATRA(5 μmol/L)刺激。培养目标时间后,每孔加入10 μl的CCK-8溶液,继续培养1 h,轻轻拍动培养板充分混匀后,在酶标仪上测定450 nm的吸光度(A),并绘制细胞生长活力曲线。
-
以每孔5×104个细胞的密度将LX-2细胞接种于24孔培养板(预置细胞爬片),培养过夜后以不同方式刺激培养48 h,以4%的多聚甲醛固定,并经Triton X-100(0.1%)透化10 min,随后经1%的BSA封闭1 h,加入一抗,4 ℃过夜孵育后,加入FITC标记的二抗,避光孵育1 h。加入DAPI染色1 min,以PBS-T缓冲液清洗后,用抗淬灭封片剂封片,于荧光倒置显微镜下观察并拍照。
-
以每孔2×105个细胞的密度将LX-2细胞接种于12孔培养板,培养过夜后以不同方式刺激培养48 h,利用RIPA试剂提取总蛋白。以20 μg总蛋白作为上样量,经SDS凝胶电泳分离后,转至甲醇预处理的PVDF膜。以5%脱脂牛奶常温封闭1 h,加入一抗,4 ℃过夜孵育后,加入辣根过氧化物酶标记二抗,常温孵育1 h。用TBS-T缓冲液清洗3次,经显色后在凝胶成像仪上观察并拍照记录。
-
以每孔2×105个细胞的密度将LX-2细胞接种于12孔培养板,培养过夜后以不同方式刺激培养48 h,利用trizol试剂提取总RNA。以200 ng总RNA为模板,逆转录成互补DNA(cDNA),反应程序为37 ℃,10 min,85 ℃,5 s。以稀释后的cDNA为模板进行实时定量PCR反应,反应程序为95 ℃,15 s;56 ℃,20 s;72 ℃,20 s,共40个循环。以GAPDH基因作为内参,每个样品重复3次,经2−△△Ct法计算目的基因的相对表达水平。
-
细胞内ROS利用DCFH-DA荧光探针检测。以每孔1×104个细胞的密度将LX-2细胞接种于24孔培养板,培养过夜后以不同方式刺激培养48 h。去除培养液并加入100 μl含DCFH-DA(10 μmol/L)的无血清培养基,继续孵育20 min。用无血清培养基清洗3次后,在荧光显微镜下观察并拍照记录。
以每孔1×105个细胞的密度将LX-2细胞接种于6孔培养板,培养过夜后以不同方式刺激培养48 h。细胞内GSH和MDA水平根据试剂盒说明书进行检测,计算总蛋白中GSH和MDA的含量(nmol/mg)。
-
双荧光自噬流通过转染自噬双标腺病毒(pAd-mRFP-GFP-LC3)后检测。以每孔1×104个细胞的密度将LX-2细胞接种于24孔培养板,培养过夜。将腺病毒转染细胞后,以不同方式刺激培养48 h,即在荧光显微镜下分别观察红色及绿色荧光信号并拍照记录。自噬激活后,自噬体与溶酶体融合后绿色荧光发生淬灭,红色荧光增强。
-
所有数据均使用SPSS 26.0软件进行统计学分析,满足正态分布的计量数据以(
$ \bar x \pm s $ )表示。组间差异以独立样本t检验比较分析,以P<0.05说明差异具有统计学意义。 -
如图1A所示,常规培养条件下,5 μmol/L的ATRA处理48 h和72 h后的HSCs生长活力为对照组的(84.5±4.8)%和(86.7±3.0)%,具有一定的抑制作用。如图1B所示,在PDGF-bb诱导条件下,5 μmol/L的ATRA处理48 h和72 h后的HSCs生长活力为对照组的(52.4±3.0)%和(57.6±2.0)%,具有显著的抑制作用(P<0.01)。
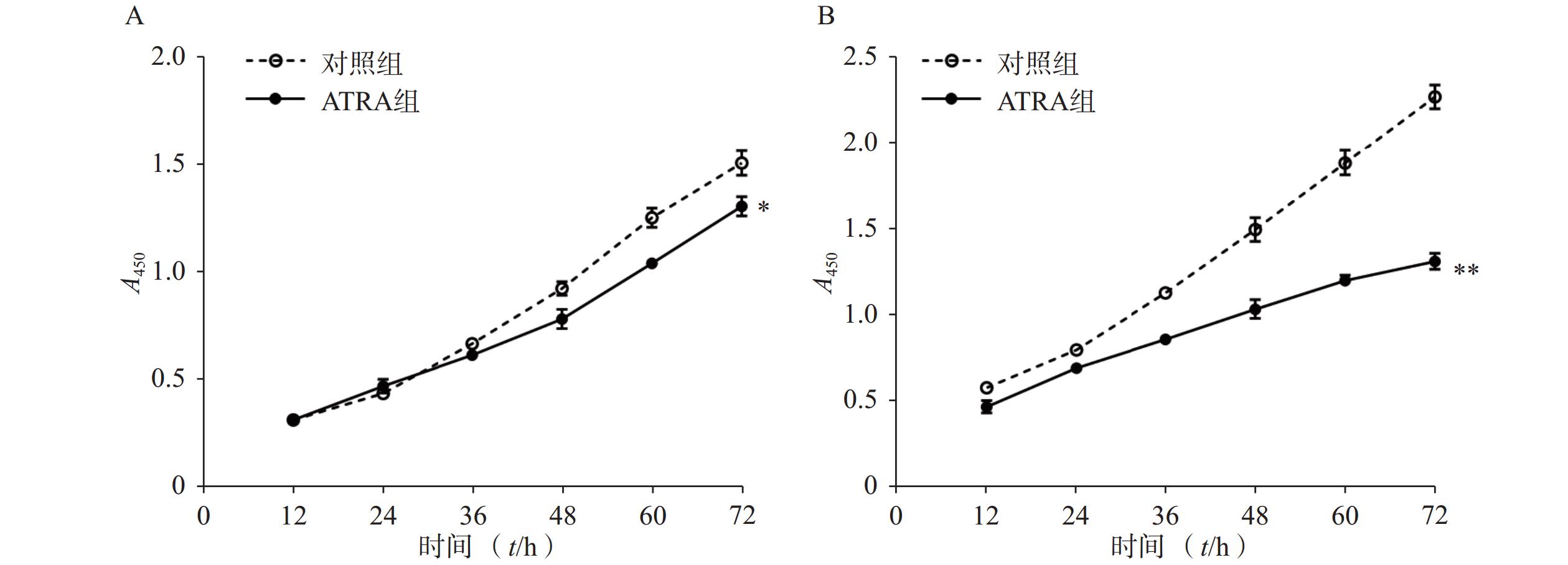
图 1 ATRA对HSCs生长活力的影响(n=5)
-
免疫荧光结果如图2A所示,与对照组相比,PDGF-bb刺激的HSCs中α-SMA的绿色荧光信号较强,而ATRA处理后,α-SMA荧光信号显著降低。蛋白质免疫印迹的结果如图2B所示,与对照组相比,PDGF-bb刺激的HSCs中α-SMA和Collagen I的蛋白表达明显增加,而ATRA处理后,α-SMA和Collagen I蛋白表达明显降低。
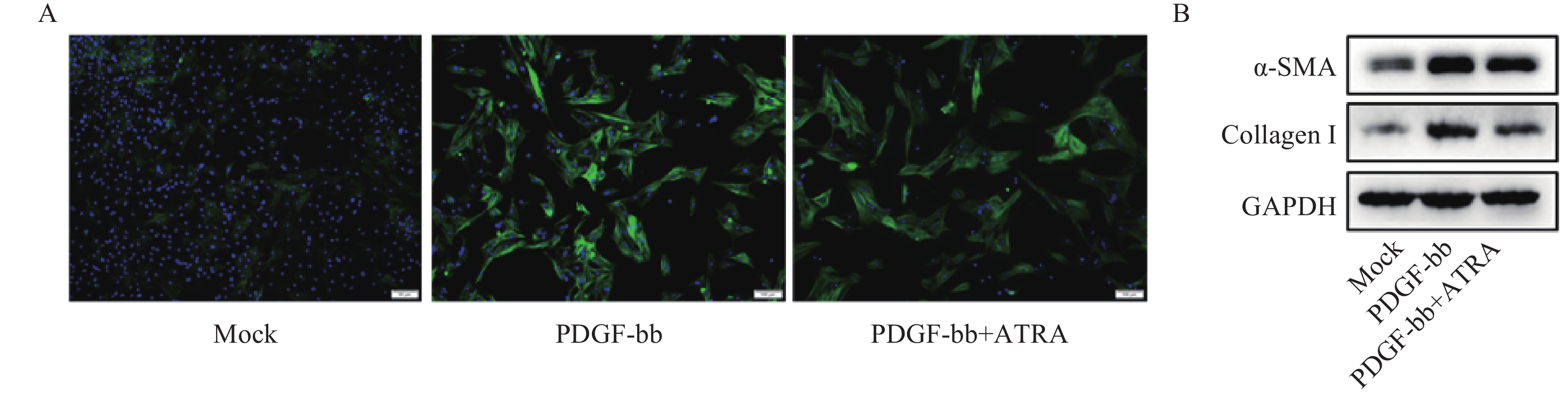
图 2 ATRA对PDGF-bb诱导HSCs活化的影响
-
ROS的检测如图3A所示,PDGF-bb刺激后HSCs中的荧光强度明显强于对照组,而ATRA处理后荧光强度明显降低,提示ATRA抑制HSCs中ROS的生成。如图3B所示,PDGF-bb刺激后,HSCs中GSH的含量明显低于对照组(18.82±1.83 nmol/mg vs. 46.45±1.69 nmol/mg),MDA的含量则明显高于对照组(13.46±1.43 nmol/mg vs. 5.45±0.47 nmol/mg);ATRA处理后GSH的含量明显升高(32.60±2.23)nmol/mg,MDA的含量则明显降低(9.56±0.34)nmol/mg。
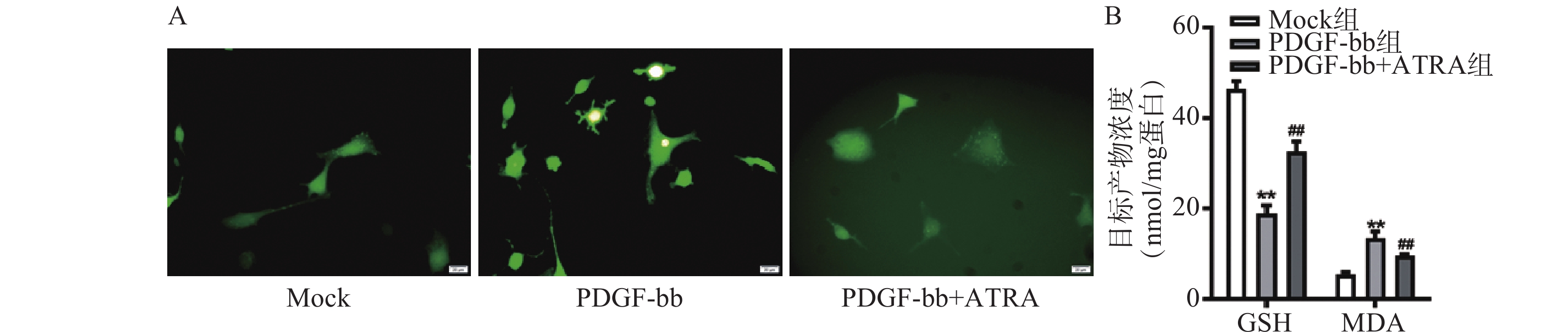
图 3 ATRA对HSCs氧化应激的影响
-
实时定量PCR的检测结果如图4A所示,ATRA处理后HSCs中抗氧化基因NRF2、HO-1和ATF4的表达明显增加(P<0.01),分别是PDGF-bb组的(2.53±0.15)倍、(3.34±0.12)倍和(2.58±0.10)倍。蛋白质免疫印迹的结果如图4B所示,NRF2、HO-1和ATF4的蛋白表达在ATRA处理后也明显增加。
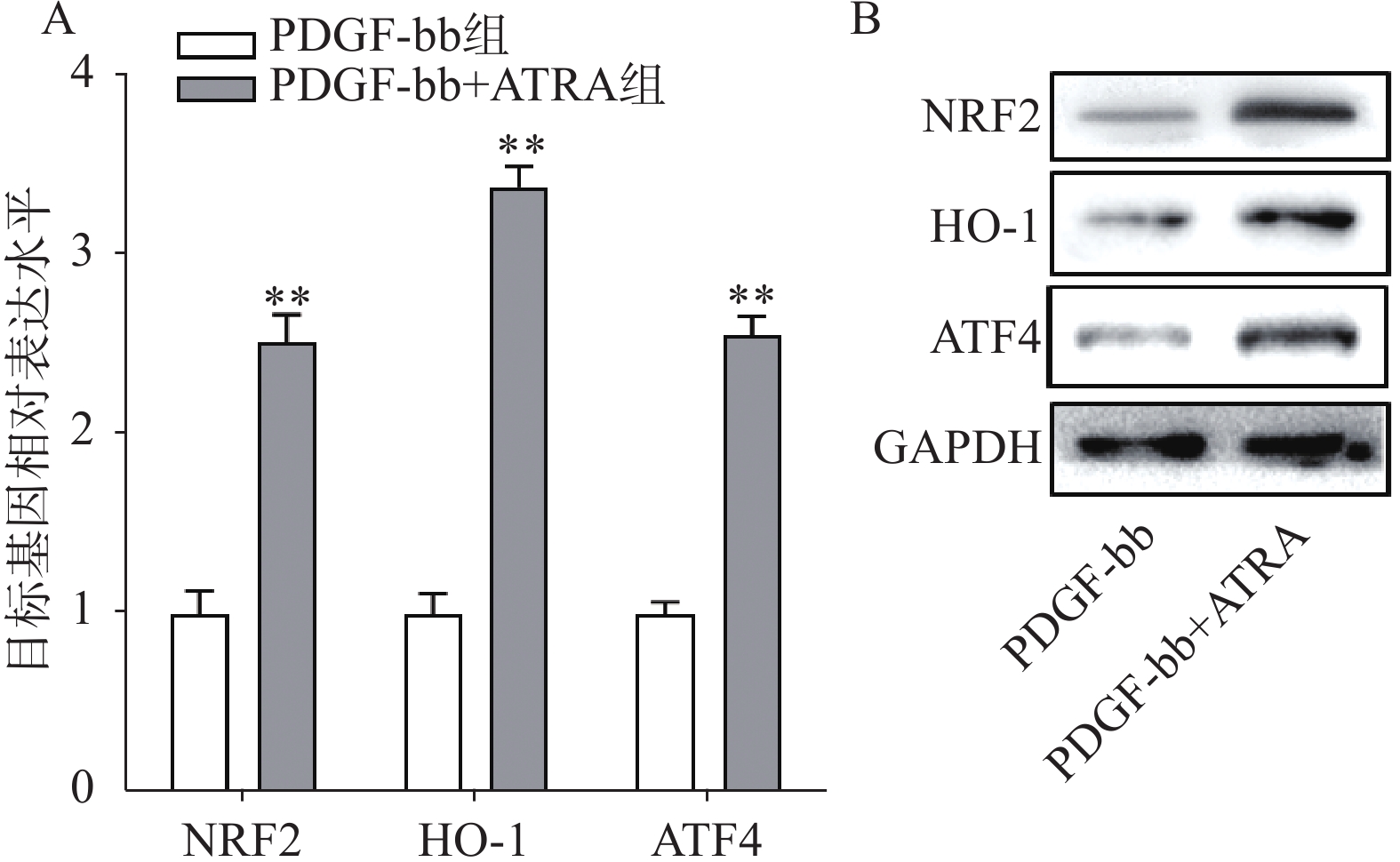
图 4 ATRA对HSCs中抗氧化基因表达的影响
-
蛋白质免疫印迹的结果如图5A所示,ATRA处理后HSCs中自噬标志蛋白Beclin 1的表达和LC3 II/I均明显减少。双荧光自噬流的检测结果如图5B所示,ATRA处理后HSCs中红色荧光信号显著降低,绿色荧光信号明显增强,自噬流信号显著降低。
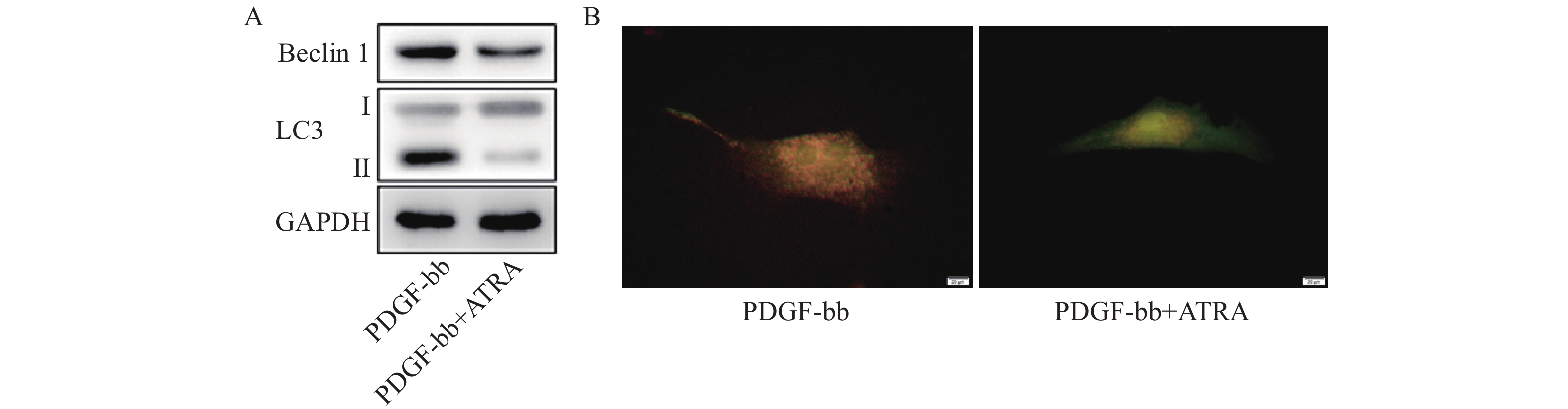
图 5 ATRA对HSCs自噬活力的影响
-
目前,还没有特定的抗纤维化疗法来预防或逆转肝纤维化,现有的治疗方案旨在去除潜在的致病因子或紊乱,但也证明了肝纤维化的潜在可逆性[7]。研究证实,HSCs是主要的肝胶原生成细胞,并被认为是肝纤维化的主要效应细胞[8]。在健康肝脏中,HSCs位于窦周间隙,处于静止状态;当受到损伤或刺激时,HSCs会对各种促纤维化信号做出反应,如来自受损肝细胞的产物、来自Kupffer细胞的生长因子和细胞因子、重构的ECM以及肝外信号等[9]。HSCs的活化涉及多个进程,包括细胞死亡、衰老和恢复静止状态等。在此过程中,HSCs会失去其特有的细胞质脂滴,并转分化为肌成纤维样细胞,并合成ECM成分(如胶原纤维I型和III型),增殖、收缩和迁移等能力增强,并具有促炎作用。HSCs活化的作用机制和干预是肝纤维化防治研究的重要内容。
静息状态HSCs中富含的脂滴,其主要成分是甘油三酯和维生素A。脂滴脱落被认为是HSCs活化的形态学标志,但其生物学作用仍被广泛研究[10-12],作为维生素A的主要代谢产物,ATRA被发现广泛参与肝纤维化以及肝癌等的生物学过程[13, 14]。研究发现,ATRA能通过抑制硫氧还蛋白互用蛋白的表达,改善TGF-β诱导的HSCs的活化和维生素A缺乏诱导的肝纤维化[6]。目前的研究证实,ATRA 通过与 RAR(α、β、γ)或 PPARβ/δ 起作用,但这两组受体的生物效应在某些进程中却完全相反[15],提示ATRA抗肝纤维化的作用机制未完全阐明。还有研究发现,纤维化药物对HSCs的激活最终会导致这些细胞的衰老,尽管有助于纤维化的逆转,但也可能会导致肝癌的发生[16]。因此,ATRA作为抗肝纤维化潜在药物的应用潜能仍需深入探索和验证。
研究证实,氧化应激与肝纤维化之间存在密切关系,当肝脏受到氧自由基的攻击时,机体抗氧化防御系统的平衡就会被打破,从而导致氧化应激[17, 18]。MDA是脂质过氧化的主要代谢产物,过量时会严重破坏细胞膜结构,也被认为是肝脏中自由基的间接指标[19]。GSH是哺乳动物细胞中最主要的自由基清除剂,广泛分布于包括肝脏在内的多个器官。研究发现,GSH可保护肝细胞免受各种自由基的侵害,包括ROS、脂质氢过氧化物、异生物毒物和重金属[20]。因此,调节MDA和GSH水平有助于控制氧化应激,最终可能有助于抑制肝纤维化。有研究报道,抑制氧化应激显著降低MDA水平,提高GSH水平,对CCl4诱导的小鼠肝脏纤维化具有明显的保护作用[21]。本课题研究发现ATRA处理促进了HSCs中抗氧化基因的表达,造成细胞中GSH的增加和MDA的降低,减少细胞内ROS的水平,缓解了PDGF-bb诱导的HSCs氧化应激,研究结果提示,以氧化应激为靶点可能是肝纤维化防治的潜在策略。
自噬是真核细胞消除一次性或有潜在危险的细胞质物质的一种新陈代谢过程,可以消除有缺陷的蛋白质和细胞器、细胞内的病原体,防止异常蛋白质的积累,在许多疾病的病理过程中发挥着积极作用[22]。越来越多的证据表明,肝细胞和非实质性细胞(HSCs、Kupffer 细胞等)的自噬反应对肝脏的生理功能至关重要[23]。近年来,HSCs自噬成为肝纤维化研究领域的热点,其调控机制日益受到关注。研究显示,自噬水平的增加可加速HSCs中脂滴的降解,为HSCs的激活提供能量支持[24, 25]。降低HSCs自噬活性显著抑制其活化,降低小鼠的肝纤维化程度[26]。本课题的结果证实,ATRA能抵抗PDGF-bb诱导的HSCs自噬水平的增加,这可能是ATRA抑制HSCs活化的分子机制之一。
HSCs是目前研究药物代谢和毒理的重要工具,也是研究肝纤维化的绝佳模型[27]。HSCs具有高度的可塑性,在有害刺激下会改变其表型和代谢,并产生大量ECM成分来替代受伤细胞,生成纤维化疤痕[28]。在各种HSCs细胞系中,LX-2是目前广泛使用的细胞类型[29]。研究发现,LX-2细胞会根据培养基中FBS的浓度改变其代谢,从而诱导基因的差异表达。在低浓度FBS下,LX-2细胞呈静止样表型;在高浓度 FBS下,细胞呈肌成纤维细胞样表型,产生ECM成分[30]。本课题研究发现,PDGF-bb可诱导LX-2细胞向肌成纤维细胞样表型分化,表现为高增殖水平,且高表达α-SMA和Collagen I,而ATRA的处理显著降低细胞增殖能力并抑制α-SMA和Collagen I的表达,表明ATRA具有抵抗HSCs活化的潜在功能。
本课题在细胞水平探索了ATRA对HSCs激活的作用和潜在机制。ATRA促进抗氧化基因的表达,降低PDGF-bb诱导的HSCs氧化应激及自噬活力,抑制了HSCs的激活。研究结果证实了ATRA对肝纤维化防治的潜在应用。需要注意的是,自噬活性的激活存在“双刃剑”作用,ATRA对HSCs自噬活性的调节还需要进行剂量和分子机制等方面的深入研究。
Exploration of the role and mechanism of all-trans retinoic acid on activation and oxidative stress of hepatic stellate cell
-
摘要:
目的 探讨全反式维甲酸(ATRA)对肝星状细胞(HSCs)活化及氧化应激的作用和潜在机制。 方法 应用10 ng/ml的血小板源性生长因子(PDGF-bb)诱导HSCs活化,以5 μmol/L剂量的ATRA处理48 h。检测细胞生长活力和表型标志物表达的变化,评价ATRA对HSCs活化的影响;检测细胞内活性氧(ROS)、还原型谷胱甘肽(GSH)和丙二醛(MDA)、抗氧化基因表达的变化,评价ATRA对HSCs氧化应激的影响;检测自噬标志物表达和自噬流的变化,评价ATRA对HSCs自噬活性的影响。 结果 与PDGF-bb组相比,ATRA处理的HSCs生长活力显著降低(P<0.01),α-SMA和Collagen I蛋白的表达明显减少(P<0.01),细胞内ROS和MDA显著减少(P<0.01),GSH显著增加(P<0.01),抗氧化基因NRF2、HO-1和ATF4的表达明显增加(P<0.01);同时自噬标志物Beclin 1和LC3 II/I的表达明显减少(P<0.01),自噬流信号显著降低。 结论 ATRA显著抑制PDGF-bb诱导的HSCs活化,降低HSCs的氧化应激水平和自噬活性,对肝纤维化的防治具有潜在应用价值。 Abstract:Objective To explore the role and potential mechanisms of all-trans retinoic acid (ATRA) on activation and oxidative stress of hepatic stellate cell (HSC). Methods Platelet-derived growth factor (PDGF-bb, 10 ng/ml) was applied to induce the activation of HSCs, which was then treated with ATRA at a dosage of 5 μmol/L for 48 h. The effects of ATRA on HSC activation were evaluated by detecting changes in cell growth viability and phenotypic marker expression. The effects of ATRA on HSC oxidative stress were evaluated by detecting changes in intracellular reactive oxygen species (ROS), reduced glutathione (GSH) and malondialdehyde (MDA), and the expression of antioxidant genes. The effects of ATRA on HSC autophagic activity were evaluated by detecting changes in autophagy marker expression and autophagic flow. Results Compared with the PDGF-bb group, the cell viability was significantly reduced in ATRA-treated HSCs (P<0.01), as well as the expression of α-SMA and Collagen I. The intracellular levels of ROS and MDA were significantly reduced in ATRA-treated HSCs (P<0.01), whereas the GSH level was significantly increased (P<0.01). The expression levels of antioxidant genes (NRF2, HO-1, and ATF4), were significantly higher in ATRA-treated HSCs than those in the normal ones under PDGF-bb condition (P<0.01). Meanwhile, the expression of autophagy markers Beclin 1 and LC3 Ⅱ/I, and signal of autophagy flow in ATRA-treated HSCs were found to be significantly reduced (P<0.01). Conclusion ATRA significantly inhibited PDGF-bb-induced HSC activation and reduced the level of oxidative stress and autophagic activity of HSCs, which had potential applications in the prevention and treatment of liver fibrosis. -
Key words:
- all-trans retinoic acid /
- hepatic stellate cells /
- activation /
- oxidative stress /
- autophagy
-
神经损伤是世界范围内致残和导致死亡的主要原因,神经损伤疾病患病率的大幅增加导致了全社会的健康负担加重[1]。神经损伤主要包括颅脑损伤(TBI)和缺血性脑卒中(CI)等,其中TBI是最常见的神经损伤类型[2],分为急性和慢性两个阶段,炎症是这两个阶段的共同特征,目前还没有有效治疗TBI的药物和方法,迫切需要寻找具有更广泛作用的药物,以缓解TBI后炎症反应[3]。脑缺血是神经损伤患者死亡的最常见病因之一[4],大脑因供血中断而致脑缺血,进而导致中风等神经损伤性疾病[5]。CI诱导了多种细胞死亡形式,如兴奋性毒性、酸毒性和离子失衡、氧化/氧化应激、炎症[6]、凋亡和梗死周围去极化等。目前CI最有效的治疗手段是静脉溶栓和血管内取栓以达到快速再灌注,这两者都能降低患者致残率,但都需要在发病4小时内尽快完成[4],目前尚无有效的药物治疗CI患者。
中药具有多途径、多靶点的特点,已有2000多年的发展历史和临床用药经验[7],如唐·孙思邈所著《备急千金方》中“小续命汤治卒中风欲死”和“大秦艽汤(金·刘完素)治中风”等。因此,阐明在临床上广泛使用的中药的作用机制是本文关注的重点。益母草来自唇形科植物益母草(Leonurus japonicus Houtt)的新鲜或干燥地上部分,临床上主要用于子宫收缩和镇静[8]。现代药理学研究发现其具有子宫收缩、抗炎、镇痛和抗氧化作用等[9]。值得引起关注的是,益母草对神经损伤也有保护作用[10]。然而,关于益母草治疗神经损伤的物质基础和作用机制的研究还未见报道。因此有必要阐明益母草治疗神经损伤的物质基础和作用机制。
本研究旨在利用网络药理学预测益母草的活性成分、靶点及相关通路来探讨其治疗神经损伤的潜在分子机制,为益母草的药理机制深入研究和临床应用提供参考。
1. 材料与方法
1.1 数据库及软件
中药系统药理学分析平台(TCMSP)和中药分子机制的生物信息学分析工具(BATMAN-TCM);活性成分靶标预测数据库(SwissTargetPrediction, STP);人类基因注释数据库(GeneCards);疾病靶点标准化数据库(Uniprot);京都基因与基因组百科全书; 蛋白-蛋白相互作用网络平台(STRING 11.0);Venny2.1软件、Cytoscape 3.6.0软件和在线作图工具微生信。
1.2 益母草活性成分的筛选
通过TCMSP和BATMAN-TCM数据库输入“yimucao”,搜索得到益母草的活性成分, 然后,在TCMSP中设置口服生物利用度(OB)≥30%及药物相似性(DL)≥0.18;在BATMAN-TCM中设置“药物-靶点”相似性模型阀值≥20,调节P值≤0.05,筛选活性成分。
1.3 活性成分靶点的预测和筛选
在获得益母草活性成分的基础上,检索TCMSP和STP数据库,限定种属为“Homo sapiens(人类)”,获取活性成分的作用靶点。通过PubChem数据库来确证收集到的活性成分,将其标准化并下载 SMILES序列。再通过TCMSP数据库搜索确证后的活性成分的靶点,将搜索的靶点按照度值从大到小排列后得到益母草的潜在靶点。在数据库STP中,搜索SMILE式,筛选条件为“概率>0”,删除重复值后预测得到药物的潜在靶点。此外,由于益母草中葫芦巴碱已被证实具有较好的治疗神经损伤作用,故将该化合物也纳入活性成分范围内[11],并通过TCMSP和STP数据库获取其成分靶点。
1.4 疾病相关靶点的筛选
通过GeneCards、DisGenet、OMIM数据库以疾病名称“cerebral ischemia”和“traumatic brain injury”进行检索,获得神经损伤相关靶标。
1.5 药物靶点与疾病靶点标准化及PPI网络构建
利用疾病靶点标准化数据库Uniprot,分别上传上述得到的益母草潜在靶点与神经损伤相关靶点名,获取其靶点的标准基因名以及Uniprot ID。为明确益母草治疗神经损伤疾病的潜在靶点,将二者的靶点上传至Venny 2.1 软件绘制韦恩图,并导出交集的基因。再将筛选得到的共有靶标蛋白上传至STRING平台,选择“multiple proteins”模式,建立药物靶蛋白-疾病靶蛋白相互作用网络,结合分值取中等“medium confidence(≥0.4)”,其余参数默认,利用 Cytoscape3.6.0软件构建益母草治疗神经损伤的PPI网络。利用cytoHubba插件计算 PPI网络每个节点的度值,筛选益母草治疗神经损伤的核心靶点。
1.6 富集分析
在上述STRING中的结果下,选择“Analysis”,点击下载“Biological Process(GO)”“Molecular Function(GO)”“Cellular Component(GO)”“KEGG Pathways”。阈值设置为P≤0.01, 并按照涉及的靶点数目多少进行排序,得到GO气泡图和 KEGG 信号通路条形图。
2. 结果
2.1 益母草活性成分的筛选
通过TCMSP数据库检索到益母草已报道的化学成分,以ADME参数OB≥30%、DL≥0.18进行筛选,得到益母草活性成分8个;BATMAN-TCM数据库检索到10个益母草活性成分;加上文献检索的1个化合物共19个(表1)。再将这19个活性成分输入TCMSP以及STP数据库,搜索的结果经过筛选去除重复项后共得到654个益母草潜在靶点。
表 1 益母草筛选所得活性成分序号 化合物 来源数据库 1 没食子酸 BATMAN-TCM 2 水苏糖 BATMAN-TCM 3 芦丁 BATMAN-TCM 4 月桂酸 BATMAN-TCM 5 水苏碱 BATMAN-TCM 6 益母草素 BATMAN-TCM 7 西班牙夏罗草酮 BATMAN-TCM 8 鸟嘌呤 BATMAN-TCM 9 益母草碱 BATMAN-TCM 10 4-胍基丁醇 BATMAN-TCM 11 鼬瓣花二萜 TCMSP 12 ZINC04073977 TCMSP 13 前益母草二萜 TCMSP 14 异前益母草二萜 TCMSP 15 槲皮苷 TCMSP 16 花生四烯酸 TCMSP 17 异鼠李素 TCMSP 18 山奈酚 TCMSP 19 葫芦巴碱 文献 2.2 疾病靶点的筛选
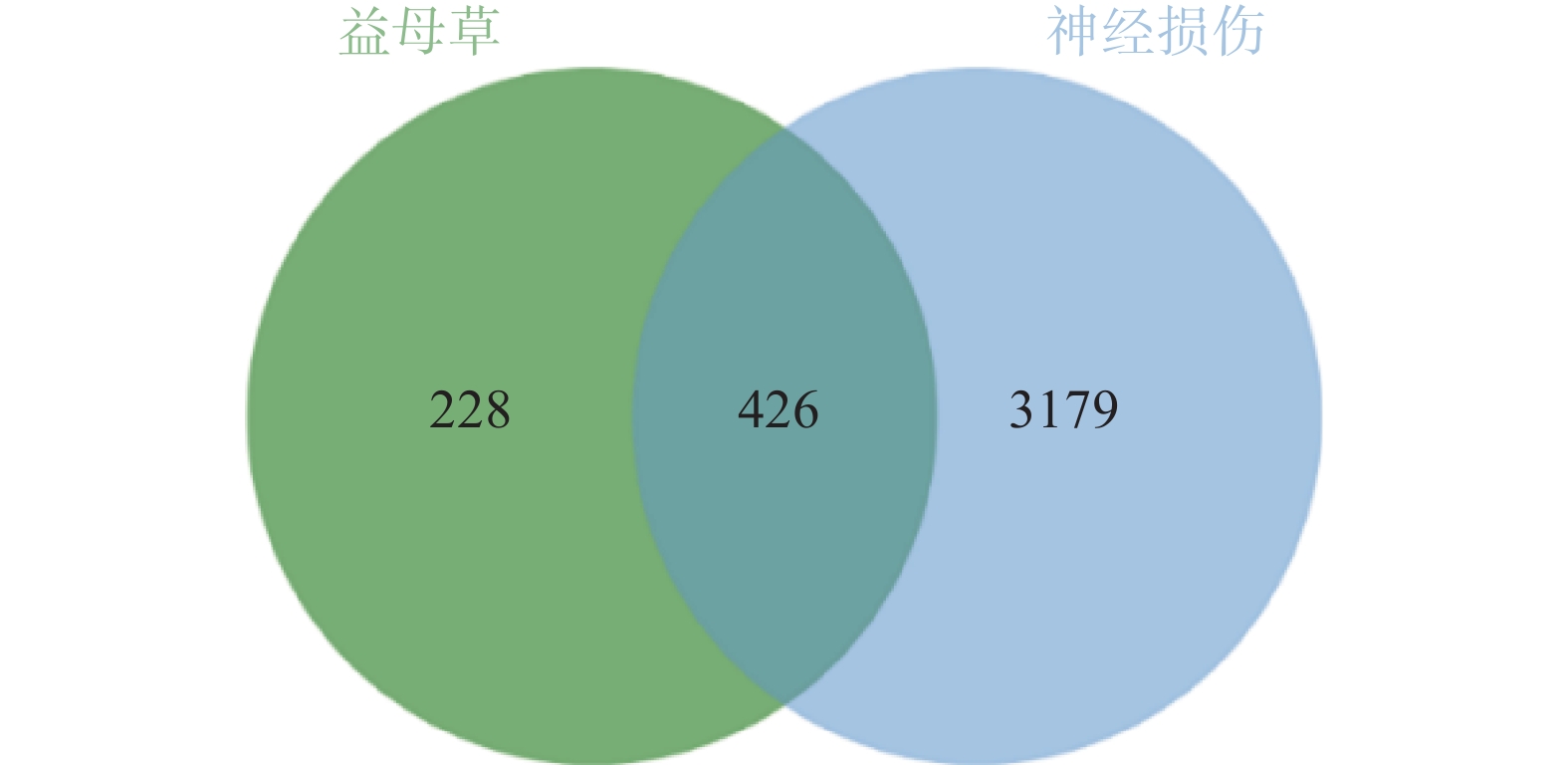
通过GeneCards、DisGenet和OMIM数据库以疾病名称为“cerebral ischemia”和“traumatic brain injury”分别进行检索,在DisGenet数据库中没有检测到TBI靶点,删除重复值后,得到神经损伤的靶点3605个,将疾病相关的靶点与益母草靶点进行Venn交集分析,筛选得到益母草治疗神经损伤的潜在靶点426个,并获得药物-疾病共同靶点基因韦恩图(图1)。
2.3 PPI 网络分析
将益母草治疗神经损伤的426个潜在靶点,导入STRING数据库,将相互作用靶点的结果导入Cytoscape 3.6.0进行可视化分析,得到由331个节点、6955条边共同组成的网络(图2),同时得到网络中关键靶点的度值(表2)。如图2所示,与神经损伤相关度较高的靶点(度值≥139)为丝氨酸/苏氨酸蛋白激酶1(AKT1)、白细胞介素6(IL-6)受体、血管内皮生长因子A(VEGFA)、半胱氨酸蛋白酶3(CASP3)、肿瘤蛋白P53(TP53)、基质金属蛋白酶-9 (MMP9)。度值大的靶点提示在网络调控中起关键作用,这些度值大的靶点很可能是益母草治疗神经损伤的关键靶点。
表 2 益母草治疗神经损伤相关靶点信息基因 度值 靶点名称 数据库中代码 AKT1 225 丝氨酸/苏氨酸蛋白激酶 P31749 IL6 217 白介素6 P05231 VEGFA 196 血管内皮生长因子A P15692 TNF 187 肿瘤生长因子 P01375 TP53 186 细胞肿瘤抗原P53 P04637 SRC 165 原癌基因酪氨酸受体激酶 P12931 CASP3 163 胱天蛋白酶-3 P42574 MAPK1 160 丝裂原活化蛋白激酶1 P28482 CXCL8 157 白介素8 P10145 EGFR 153 表皮生长因子受体 P00533 EGF 150 前表皮生长因子 P01133 PTGS2 146 牛前列腺素G/H合成酶2 P35354 MAPK8 146 丝裂原活化蛋白激酶8 P45983 MYC 146 原癌基因蛋白Myc P01106 JUN 145 转录因子AP-1 P05412 STAT3 143 信号传导及转录激活子3 P40763 FOS 143 原癌基因c-Fos P01100 MMP9 139 基质金属蛋白酶9 P14780 IL-1β 133 白介素1β P01584 2.4 GO生物过程和KEGG通路富集分析
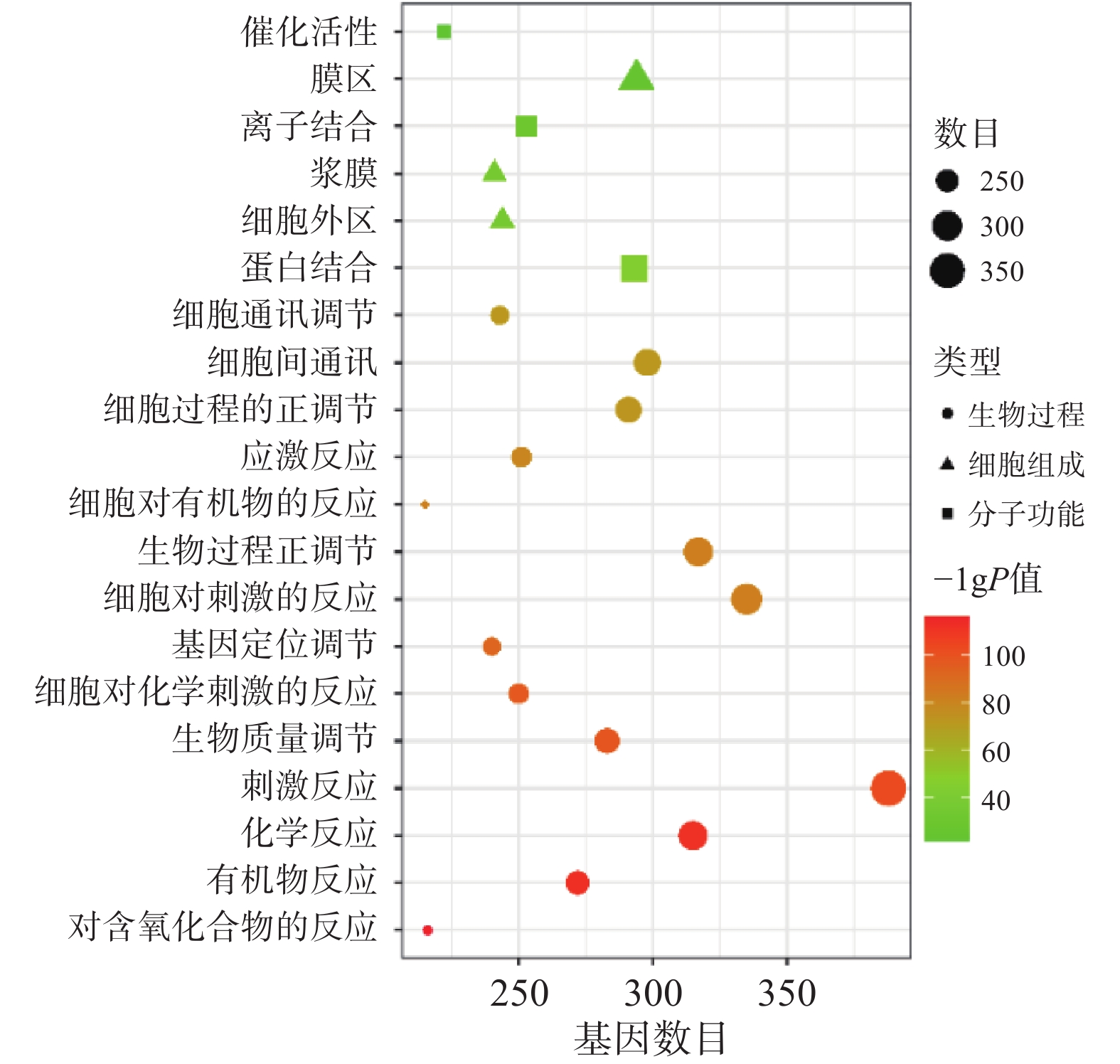
将“2.2” 项下获取的426个潜在的治疗神经损伤的靶点通过STRING进行生物过程(BP)、细胞组分(CC)和分子功能(MF)分析,以 P<0.01为条件,筛选靠前的GO富集分析,如图3所示。图中纵坐标表示富集条目,横坐标表示基因计数,颜色深浅代表-log10(p)值大小。其中 GO-BP 主要为应激反应、生物调节和细胞通讯等;GO-CC主要为细胞膜等;GO-MF主要为蛋白质结合、离子结合和催化还原活性等。
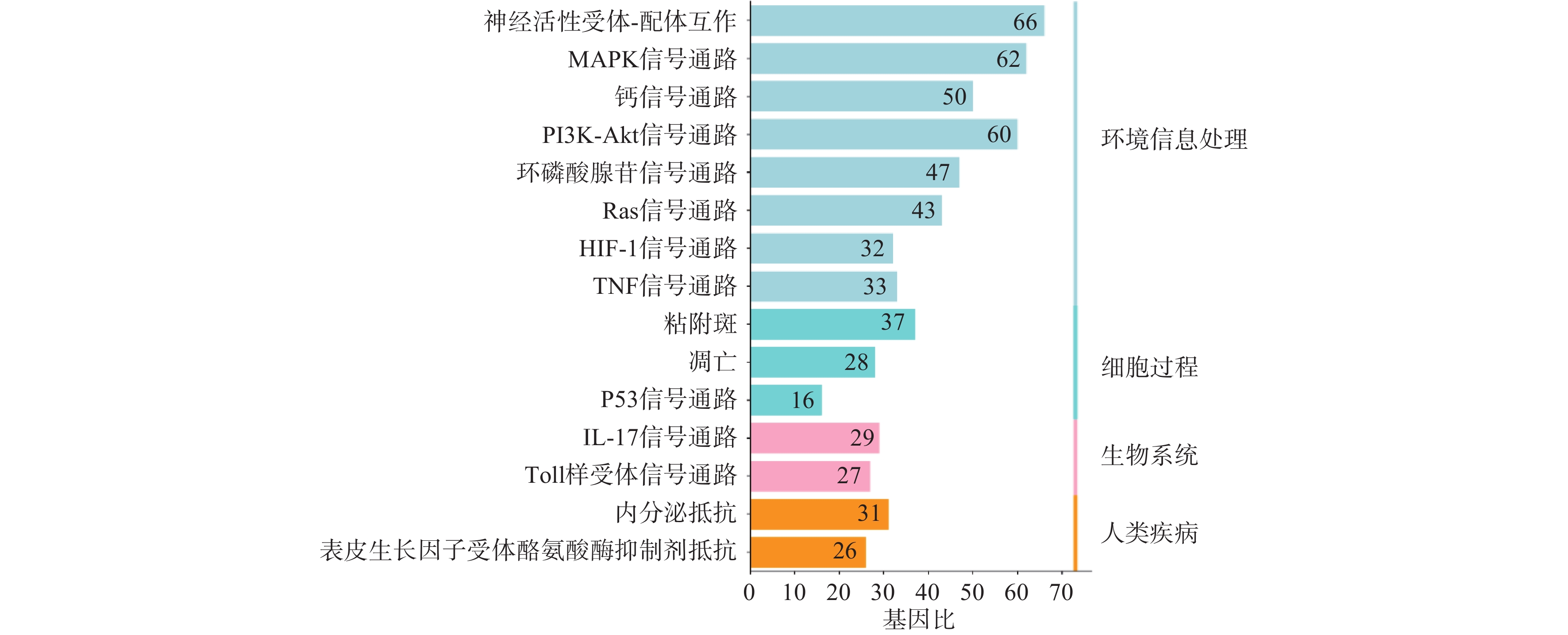
利用STRING数据库对益母草治疗神经损伤的潜在的426个靶点进行富集分析,筛选出显著的前16条信号通路(P<0.01),主要涉及的信号通路为MAPK、Toll样受体、PI3K-Akt、肿瘤坏死因子、IL-17和凋亡等信号通路(图4)。
2.5 药物“活性成分-靶点”网络的构建与分析
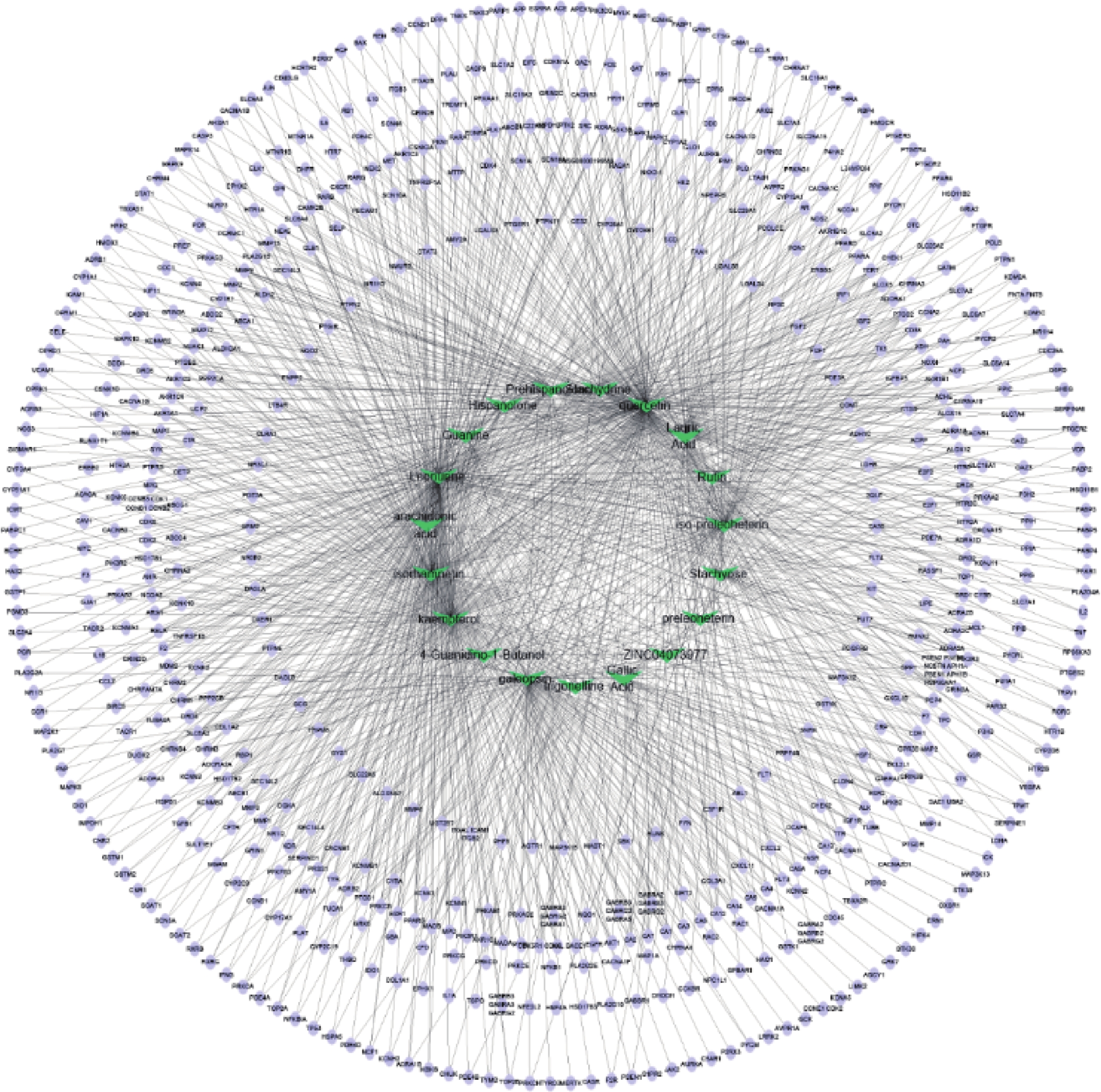
通过Cytoscape3.6.0软件得到药物“活性成分-靶点”的网络(图5)。网络中化合物19个,靶点426个。在图5中,箭头节点代表益母草中化合物,椭圆形节点代表靶点,结果可以明显看出益母草中度值相对较高的化合物有槲皮素、益母草碱、山奈酚、异鼠李素、水苏碱、葫芦巴碱等,这些化合物可能是益母草治疗神经损伤的关键化合物。
3. 讨论
本研究采用网络药理学方法,借助相关数据库以及各种绘图软件对益母草治疗神经损伤的物质基础和作用机制进行研究。共筛选出益母草有效活性成分19个,对应活性成分靶点654个。其中,益母草与神经损伤的共同基因有426个,“药物活性成分-共同靶点”网络与PPI网络结合分析,结果发现,益母草治疗神经损伤的关键活性成分有槲皮素、益母草碱、山奈酚、异鼠李素、水苏碱、葫芦巴碱等,其中槲皮素对脑缺血的作用最为突出。槲皮素通过发挥抗氧化[12]、抗炎和抗凋亡作用[13]对脑缺血的病理学改变产生了积极的治疗作用。益母草碱通过抗氧化、抗凋亡、保护线粒体和激活Nrf-2/HO-1信号通路发挥血脑屏障保护作用[14]。山萘酚具有抗氧化、抗炎、抗癌和预防心血管疾病等多种药理活性[15]。异鼠李素具有保护心脑血管、抗肿瘤、抗炎、抗氧化、保护器官、预防肥胖等作用[16]。水苏碱通过多种分子机制(包括TGF-β、ers介导的细胞凋亡、MMPs/TIMPs、NF-κB和JAK/STAT)抑制细胞外基质(ECM)沉积、降低炎症和氧化应激,以及血管生成保护血管等发挥神经保护作用[17]。葫芦巴碱具有抗凋亡、抗炎、抗氧化、神经保护等多种药理作用,具有改善认知能力的潜力。
根据益母草靶蛋白互作网络图分析可知,益母草治疗神经损伤的核心靶点主要为:AKT1,IL-6,VEGFA,CASP3, TP53,MMP-9。AKT1能够通过丝氨酸和/或苏氨酸磷酸化介导调节细胞代谢、增殖、细胞存活、胰岛素信号传导、生长和血管生成过程。据报道[18]阿托伐他汀通过抑制JNK3/cJun/caspase-3,增强Akt-nNOS信号通路,抑制脑缺血大鼠脑内细胞凋亡,对脑缺血再灌注有保护作用。IL-6可调节多种细胞的生长与分化,具有调节免疫应答、急性期反应及造血功能,并在机体的抗感染免疫反应中起重要作用。IL-6作为促炎细胞因子在脑缺血中的作用可能是通过NF-κB通路来实现的[19]。据报道,LncRNA MEG8通过miR-130a-5p/VEGFA信号靶向减轻缺血性中风后的脑缺血[20],通过靶向VEGFA,下调microRNA-195促进血管生成[21]。Caspase-3(CASP3)是细胞凋亡途径中最关键的酶类之一,与癌症的发生、衰老、心脑血管疾病的发生等有着重要联系。Nahid等[22]研究发现通过降低Bax/Bcl-2比值和caspase-3活化,可减轻脑缺血后海马CA1神经元损伤,改善脑缺血损伤引起的功能和记忆丧失。TP53是神经元凋亡的主要调节因子,任何降低TP53稳定性及其向线粒体迁移的方法都可以减轻缺血性脑区的神经元损失[23]。MMP-9是一种明胶酶,大脑中许多细胞都能分泌MMP-9。MMP-9降解细胞外基质成分,从而引发中风,Zinnhardt等[24]研究发现脑缺血的发生会促进基质金属蛋白酶(MMPs)的产生,尤其是MMP-9,另外MMP-9的激活又可引起血脑屏障受损。
GO功能富集分析发现,益母草治疗神经损伤的基因功能主要体现在生物调节、氧化应激反应、细胞通讯等生物学过程以及蛋白质结合、离子结合和催化还原等。KEGG信号通路富集分析显示,益母草治疗神经损伤所涉及的TNF信号通路、MAPK信号通路、TP53信号通路、PI3K-Akt信号通路的P值较小,被显著富集。TNF具有促进细胞生长、分化、凋亡及诱发炎症等生物学效应。TNF-α可以激活JNK,Caspase蛋白酶和转录因子NF-kB这三条信号通路,实现其免疫调节和细胞凋亡的生物学功能,从而对脑缺血产生影响。级联p38-MAPK的转导通路位于中枢神经系统,在缺血、缺氧等条件刺激下可被激活。可通过p38 MAPK和c-Jun抑制炎症反应,对损伤后的神经有保护作用[25]。Yao等[26]发现通过抑制MAPK信号通路的激活,恢复神经功能,减轻血脑屏障通透性破坏,对脑缺血产生保护作用。缺血神经元释放的内源性配体激活TLR信号通路,导致大量炎症细胞因子TNF-α、IL-1β、iNOS的产生,从而引起脑缺血后继发性炎症损伤。TLRs介导的缺血耐受可作为预防和治疗脑缺血的重要靶点[27]。TP53是一个肿瘤抑制蛋白,调节各种各样基因的表达,包括细胞凋亡等,此外TP53可不依赖其活性,仅作为一个转录因子来引发凋亡通路。抑制NF-κB及下游TP53可显著减轻神经元自噬和凋亡,具有显著的神经保护作用。NF-κB、TP53及其介导的自噬和凋亡在脑缺血再灌注损伤恶化中也起关键作用[28]。PI3K/AKT信号通路是一条与增殖,分化和凋亡相关的信号通路[29]。
综上所述,本研究应用网络药理学的方法预测了益母草治疗神经损伤的主要活性成分和潜在分子机制,但由于所使用平台的数据收录,更新相对滞后,中药活性成分筛选条件口服生物利用度与类药性并不是唯一的标准,因而,研究预测的结果有其局限性。需要在今后的实验研究中进一步阐明和验证益母草中活性成分的作用靶点,从而完善其治疗神经损伤的有效化学成分及作用机制。
-
图 1 ATRA对HSCs生长活力的影响(n=5)
A. 常规条件下ATRA对HSCs生长活力的影响;B. PDGF-bb刺激条件下ATRA对HSCs生长活力的影响*P<0.05,**P<0.01,与对照组比较。
图 2 ATRA对PDGF-bb诱导HSCs活化的影响
A. HSCs中α-SMA(绿色)的免疫荧光检测;B. HSCs中α-SMA和Collagen I的免疫印迹检测注A图中细胞核以DAPI(蓝色)标记,标尺为100 μm,B图中GAPDH为内参。
图 3 ATRA对HSCs氧化应激的影响
A. HSCs中ROS的DCFH-DA荧光探针检测(标尺为20 μm);B. HSCs中GSH和MDA含量的检测(n=3)**P<0.01,与Mock组比较;##P<0.01,与PDGF-bb组比较。
图 4 ATRA对HSCs中抗氧化基因表达的影响
A. HSCs中抗氧化基因的实时定量PCR检测(n=3);B. HSCs中抗氧化蛋白的免疫印迹检测**P<0.01,与PDGF-bb组比较。GAPDH为内参。
-
[1] ZUO T, XIE Q, LIU J F, et al. Macrophage-derived cathepsin S remodels the extracellular matrix to promote liver fibrogenesis[J]. Gastroenterology, 2023, 165(3): 746-761. e16. [2] COLL M, ARIÑO S, MARTÍNEZ-SÁNCHEZ C, et al. Ductular reaction promotes intrahepatic angiogenesis through Slit2-Round about 1 signaling[J]. Hepatology, 2022, 75(2):353-368. doi: 10.1002/hep.32140 [3] CHOUDHURY A, RATNA A, LIM A, et al. Loss of heat shock factor 1 promotes hepatic stellate cell activation and drives liver fibrosis[J]. Hepatol Commun, 2022, 6(10):2781-2797. doi: 10.1002/hep4.2058 [4] YU X J, ELFIMOVA N, MÜLLER M, et al. Autophagy-related activation of hepatic stellate cells reduces cellular miR-29a by promoting its vesicular secretion[J]. Cell Mol Gastroenterol Hepatol, 2022, 13(6):1701-1716. doi: 10.1016/j.jcmgh.2022.02.013 [5] WU S S, LI S F, JIN P, et al. Interplay between hypertriglyceridemia and acute promyelocytic leukemia mediated by the cooperation of peroxisome proliferator-activated receptor-α with the PML/RAR α fusion protein on super-enhancers[J]. Haematologica, 2022, 107(11):2589-2600. doi: 10.3324/haematol.2021.280147 [6] SHIMIZU H, TSUBOTA T, KANKI K, et al. All-trans retinoic acid ameliorates hepatic stellate cell activation via suppression of thioredoxin interacting protein expression[J]. J Cell Physiol, 2018, 233(1):607-616. doi: 10.1002/jcp.25921 [7] ROCKEY D C. Liver fibrosis reversion after suppression of hepatitis B virus[J]. Clin Liver Dis, 2016, 20(4):667-679. doi: 10.1016/j.cld.2016.06.003 [8] DE MESQUITA F C, GUIXÉ-MUNTET S, FERNÁNDEZ-IGLESIAS A, et al. Liraglutide improves liver microvascular dysfunction in cirrhosis: evidence from translational studies[J]. Sci Rep, 2017, 7(1):3255. doi: 10.1038/s41598-017-02866-y [9] LEE Y A, WALLACE M C, FRIEDMAN S L. Pathobiology of liver fibrosis: a translational success story[J]. Gut, 2015, 64(5):830-841. doi: 10.1136/gutjnl-2014-306842 [10] SCORLETTI E, CARR R M. A new perspective on NAFLD: focusing on lipid droplets[J]. J Hepatol, 2022, 76(4):934-945. doi: 10.1016/j.jhep.2021.11.009 [11] KLUWE J, WONGSIRIROJ N, TROEGER J S, et al. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis[J]. Gut, 2011, 60(9):1260-1268. doi: 10.1136/gut.2010.209551 [12] SENOO H, MEZAKI Y, FUJIWARA M. The stellate cell system(vitamin A-storing cell system)[J]. Anat Sci Int, 2017, 92(4):387-455. doi: 10.1007/s12565-017-0395-9 [13] LI X Y, LUO X Q, CHEN S R, et al. All-trans-retinoic acid inhibits hepatocellular carcinoma progression by targeting myeloid-derived suppressor cells and inhibiting angiogenesis[J]. Int Immunopharmacol, 2023, 121:110413. doi: 10.1016/j.intimp.2023.110413 [14] XIA S L, LIU Z M, CAI J R, et al. Liver fibrosis therapy based on biomimetic nanoparticles which deplete activated hepatic stellate cells[J]. J Control Release, 2023, 355:54-67. doi: 10.1016/j.jconrel.2023.01.052 [15] SCHUG T T, BERRY D C, SHAW N S, et al. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors[J]. Cell, 2007, 129(4):723-733. doi: 10.1016/j.cell.2007.02.050 [16] PANEBIANCO C, OBEN J A, VINCIGUERRA M, et al. Senescence in hepatic stellate cells as a mechanism of liver fibrosis reversal: a putative synergy between retinoic acid and PPAR-gamma signalings[J]. Clin Exp Med, 2017, 17(3):269-280. doi: 10.1007/s10238-016-0438-x [17] RUART M, CHAVARRIA L, CAMPRECIÓS G, et al. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury[J]. J Hepatol, 2019, 70(3):458-469. doi: 10.1016/j.jhep.2018.10.015 [18] LUANGMONKONG T, SURIGUGA S, MUTSAERS H A M, et al. Targeting oxidative stress for the treatment of liver fibrosis[J]. Rev Physiol Biochem Pharmacol, 2018, 175:71-102. [19] YANG L, BI L P, JIN L L, et al. Geniposide ameliorates liver fibrosis through reducing oxidative stress and inflammatory respose, inhibiting apoptosis and modulating overall metabolism[J]. Front Pharmacol, 2021, 12:772635. doi: 10.3389/fphar.2021.772635 [20] NARAYANANKUTTY A, JOB J T, NARAYANANKUTTY V. Glutathione, an antioxidant tripeptide: dual roles in carcinogenesis and chemoprevention[J]. Curr Protein Pept Sci, 2019, 20(9):907-917. doi: 10.2174/1389203720666190206130003 [21] GU J Y, CHEN C, WANG J, et al. Withaferin A exerts preventive effect on liver fibrosis through oxidative stress inhibition in a sirtuin 3-dependent manner[J]. Oxid Med Cell Longev, 2020, 2020:2452848. [22] HAZARI Y, BRAVO-SAN PEDRO J M, HETZ C, et al. Autophagy in hepatic adaptation to stress[J]. J Hepatol, 2020, 72(1):183-196. doi: 10.1016/j.jhep.2019.08.026 [23] UENO T, KOMATSU M. Autophagy in the liver: functions in health and disease[J]. Nat Rev Gastroenterol Hepatol, 2017, 14(3):170-184. doi: 10.1038/nrgastro.2016.185 [24] HERNÁNDEZ-GEA V, FRIEDMAN S L. Autophagy fuels tissue fibrogenesis[J]. Autophagy, 2012, 8(5):849-850. doi: 10.4161/auto.19947 [25] HERNÁNDEZ-GEA V, GHIASSI-NEJAD Z, ROZENFELD R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues[J]. Gastroenterology, 2012, 142(4):938-946. doi: 10.1053/j.gastro.2011.12.044 [26] SEKI E, BRENNER D A. Recent advancement of molecular mechanisms of liver fibrosis[J]. J Hepatobiliary Pancreat Sci, 2015, 22(7):512-518. doi: 10.1002/jhbp.245 [27] ZAHMATKESH E, OTHMAN A, BRAUN B, et al. In vitro modeling of liver fibrosis in 3D microtissues using scalable micropatterning system[J]. Arch Toxicol, 2022, 96(6):1799-1813. doi: 10.1007/s00204-022-03265-7 [28] ASRANI S K, DEVARBHAVI H, EATON J, et al. Burden of liver diseases in the world[J]. J Hepatol, 2019, 70(1):151-171. doi: 10.1016/j.jhep.2018.09.014 [29] LUCANTONI F, MARTÍNEZ-CEREZUELA A, GRUEVSKA A, et al. Understanding the implication of autophagy in the activation of hepatic stellate cells in liver fibrosis: are we there yet?[J]. J Pathol, 2021, 254(3):216-228. doi: 10.1002/path.5678 [30] XU L, HUI A Y, ALBANIS E, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis[J]. Gut, 2005, 54(1):142-151. doi: 10.1136/gut.2004.042127 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 8043
- HTML全文浏览量: 1717
- PDF下载量: 45
- 被引次数: 0

