

 下载:
下载:
 下载:
下载:

-
当归六黄汤被国家中医药管理局录入《古代经典名方目录(第一批)》[1]。其出于金元四大家李东垣编纂的《兰室秘藏·自汗门》,主治阴虚火旺之盗汗证。原方由当归、生地黄、黄芩、黄柏、黄连、熟地黄、黄芪等7味药组成。古代医家对当归六黄汤的临床应用较为单一,主要局限于汗证。而现在对当归六黄汤应用十分广泛,在原方的基础上加减药味仍取得了显著效果[2-6]。涉及上呼吸道感染、心律失常、糖尿病及其并发症、更年期综合征、甲状腺功能亢进等多个系统疾病[7-9]。
当归六黄汤的活性成分种类包括生物碱、有机酸、黄酮等,有机酸主要成分以阿魏酸为主;生物碱中主要为黄柏碱、巴马汀、黄连碱、小檗碱等;黄酮类主要有毛蕊异黄酮、黄芩素等。阿魏酸作为有机酸中含量较高的成分,具有抗肿瘤、抗氧化等药理作用[10-11],存在较大的开发潜力。黄柏碱和巴马汀均属于异喹啉生物碱类化合物,但两者极性差异相对较大。黄柏碱在降血压、抑制细胞免疫反应、中枢神经抑制方面的药理活性显著[12]。巴马汀是很多重要复方中常见的生物碱之一,极性与小檗碱相似,具有抗糖尿病和抗氧化活性等多种药学功效[13]。毛蕊异黄酮葡萄糖苷具有抗氧化、抗病毒、降血糖等多方面的药理作用[14]。该方现阶段的研究大多停留于疗效观察,物质基础、成分分析及质量控制方面的研究相对较薄弱。关于当归六黄汤的定量分析及质量控制研究目前主要采用HPLC[15]的分析方法,少量定性研究采用LC-MS[16]的分析方法。LC-MS/MS的灵敏度相对较高,可以满足对中药复方成分尤其微量成分定性定量要求以及药动学研究。该研究针对当归六黄汤主要有效成分建立了基于HPLC-QTrap的定量分析方法。采用了MRM的数据采集模式,测定当归六黄汤中4种主要成分黄柏碱、巴马汀、毛蕊异黄酮、阿魏酸的含量,以及当归六黄汤各单味药材中4种成分的含量差异,为当归六黄汤的质量控制及物质基础研究提供方法借鉴及参考依据。
-
LC-20A高效液相色谱仪(岛津);QTRAP® 4000(AB Sciex);VORTEX GENIUS 3涡旋仪(IKA);Milli-Q纯水仪(Merck Millipore);MD 200氮气吹干仪(CHINCAN);New Classic MF电子天平(万分之一,METTLER TOLEDO);Z216MK高速温控离心机(HERMLE)。
-
盐酸黄柏碱(批号:5436)、盐酸巴马汀(批号:8103)、毛蕊异黄酮(批号:7851)、阿魏酸(批号:8042)这4种对照品的质量分数均≥99%,均购自上海诗丹德标准技术服务有限公司。甲醇(质谱级,99.9%,默克);乙腈(质谱级,99.9%,默克);甲酸[色谱级,≥98%,赛默飞世尔科技(中国)有限公司];水为Milli-Q纯水仪去离子水。当归六黄汤中各单味药材均购自复旦大学附属肿瘤医院草药房,包括:当归(批号:20210615、20210705-1、20210723产自甘肃)、生地黄(批号: 20201217-1、20210324-1、20210401-1,产自河南)、熟地黄(批号:200801、210702、210801,产自河南)、黄连(批号:201101、210310、210601,产自四川)、黄柏(批号:210106、210605、210803,产自辽宁)、黄芪(批号:20200926-1、20201010-1、20210318-1,产自内蒙)、黄芩(批号:20201219-1、20210104-1、20210603-1,产自陕西)。
-
分别精密称取4个对照品(黄柏碱、巴马汀、毛蕊异黄酮、阿魏酸),用甲醇分别配制成浓度约 10 mmol/ml 的对照品溶液,取各对照品液100 μl混合均匀,加甲醇至1 ml制备成浓度为1 mmol/ml的混合对照品储备液,−20 ℃密封保存备用。
-
精密量取100 μl混合对照品储备液,首先稀释为100 μmol/ml的标准溶液,之后逐级稀释为2 μmol/ml、0.5 μmol/ml、0.2 μmol/ml、50 nmol/ml、20 nmol/ml、10 nmol/ml、5 nmol/ml、2 nmol/ml的系列标准曲线浓度。
-
样品制备方法参照文献并根据实际用量进行调整[17]:分别称取当归1 g、地黄 1 g、熟地黄1 g、黄芩1 g、黄连1 g、黄柏1 g和黄芪2 g,粉碎过20目筛后,置于250 ml圆底烧瓶中,加入10倍(V/W)体积的超纯水室温浸泡1 h,回流提取30 min,过滤收集滤液,超纯水补足体积至80 ml,随后取滤液2 ml于50 ml容量瓶,加入超纯水至刻度,获得复方供试品溶液。分别称取上述各单味药材5 g,并分别粉碎过20目筛后,按照上述方法分别进行回流提取,获得各单味药材的供试品溶液。分别取复方供试品溶液和各单味药材供试品溶液稀释10倍后进样。
-
Agilent Extend-C18色谱柱(5 μm, 4.6 mm×250 mm),流速为0.3 ml/min,进样体积为10 μl,柱温为25 ℃,流动相:0.5%甲酸水溶液(A),甲醇(B)。洗脱梯度:0 min为15% B,5 min为50% B,12~20 min为95% B,20.1~35 min 为15% B。
-
离子源为ESI源,雾化气压GS1:60 psi,辅助气压:60 psi,喷雾电压:4 500 V(正离子模式),温度:500 ℃。采集模式:多反应监测模式(MRM),阿魏酸离子对:m/z 195.3-145.1,保留时间24.03 min;巴马汀离子对:m/z 352.4-336.1,保留时间23.15 min;毛蕊异黄酮离子对:m/z 285.2-270.0,保留时间25.79 min;黄柏碱离子对:m/z 342.4-192.2,保留时间19.85 min。
-
在定量分析中,对建立的分析方法进行验证是保证测定可靠、数据可信的必要条件。方法验证主要包括以下几个方面。
-
选取20 nmol/ml浓度的混合对照品溶液进样,4种成分的总提取离子流色谱图见图1A。当归六黄汤复方供试品溶液的提取离子流色谱图见图1B。黄柏碱的提取离子流色谱图见图1C,巴马汀提取离子流色谱图见图1D,阿魏酸提取离子流色谱图见图1E,毛蕊异黄酮提取离子流色谱图见图1F。方法的专属性能够满足对中药复方主要成分的定量分析要求。
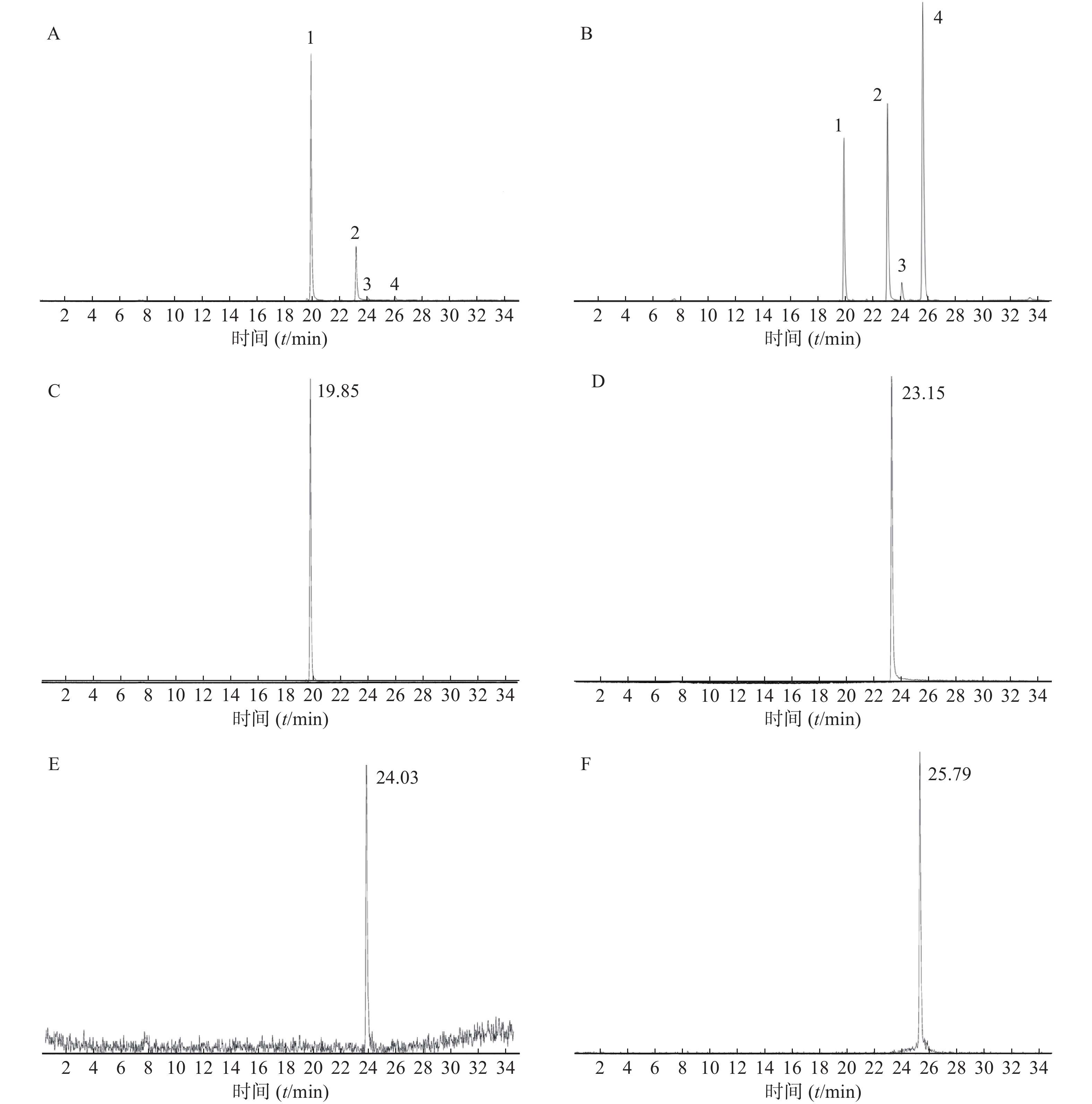
图 1 不同样品的提取离子流色谱图
-
按“2.1.2”制备一系列浓度梯度的混合对照品溶液后,由低浓度依次进样。方法的灵敏度较高,在黄柏碱浓度2 nmol/ml、巴马汀浓度20 nmol/ml、黄连碱浓度20 nmol/ml和小檗碱浓度20 nmol/ml的样品中,测得信噪比远大于10,能够满足对样品含量测定的要求。将药物峰面积和浓度之间进行线性回归,得到的标准曲线、线性范围如表1,相关系数r在 0.992 0~0.999 6之间。
表 1 4种成分的标准曲线方程、相关系数、线性范围
成分 标准曲线方程 相关系数r 浓度范围
(nmol/ml)阿魏酸 Y=443X+73.8 0.999 2 20~2 000 黄柏碱 Y=4.76×104X+1.57×104 0.995 8 2~200 巴马汀 Y=1.02×104X−4.35×103 0.996 7 20~2 000 毛蕊异黄酮 Y=1.54×103X+73.8 0.999 6 20~2 000 -
精密吸取50 nmol/ml混合对照品溶液10 μl ,连续进样6次,记录峰面积。结果显示:阿魏酸、巴马汀、毛蕊异黄酮和黄柏碱峰面积的RSD分别为1.14%、1.94%、2.09%、1.33%(n=6)。连续进样3 d,结果显示,阿魏酸、巴马汀、毛蕊异黄酮和黄柏碱峰面积的RSD分别为1.96%、2.83%、1.85%、2.15%(n=3),表明方法的精密度良好。
-
精密称取同批次当归六黄汤复方粉末6份 ,每份1.0 g,参照“2.1.3”项下方法平行制备供试品溶液后进样,测定4种成分的含量。结果显示:阿魏酸、巴马汀、毛蕊异黄酮和黄柏碱含量的RSD分别为1.89%、3.53%、2.12%、1.49%(n=6),表明方法的重复性良好。
-
分别精密吸取“2.4”项下供试品溶液,于室温放置0 h、4 h、8 h、12 h、16 h、20 h、24 h进样测定,记录峰面积。结果显示,阿魏酸、巴马汀、毛蕊异黄酮和黄柏碱峰面积的RSD分别为0.96%、1.25%、1.21%和1.07%(n=7),表明供试品溶液在室温下放置24 h内稳定。
-
取已知含量的复方供试品溶液6份,稀释100倍,每份取5 ml。分别精密加入已知浓度的对照品溶液0.5 μmol/ml阿魏酸500 μl、0.5 μmol/ml巴马汀600 μl、0.5 μmol/ml毛蕊异黄酮1 ml和0.1 μmol/ml黄柏碱500 μl混匀,加水定容至10 ml进样测定,计算加样回收率。结果显示,阿魏酸、巴马汀、毛蕊异黄酮和黄柏碱的平均加样回收率依次为97.74%、98.09%、102.50%和95.05%,RSD 依次为3.09%、2.87%、1.20%和2.14%(n=6),表明方法准确度良好,结果见表2。
表 2 4种成分的提取回收率(n=6)
成分 序号 已知浓度
(nmol/ml)加入浓度
(nmol/ml)测得浓度
(nmol/ml)提取
回收率
(%)RSD
(%)阿魏酸 1 22.50 25 46.82 97.74 3.09 2 24.17 25 50.06 3 18.10 25 42.54 4 17.83 25 41.33 5 23.63 25 48.26 6 19.84 25 43.67 巴马汀 1 32.81 30 63.57 98.09 2.87 2 36.29 30 65.61 3 32.74 30 61.52 4 34.18 30 63.13 5 33.42 30 61.83 6 30.92 30 61.25 毛蕊异黄酮 1 50.82 50 101.74 102.50 1.20 2 53.50 50 103.86 3 51.22 50 103.12 4 49.31 50 100.53 5 51.08 50 103.24 6 55.33 50 106.26 黄柏碱 1 6.54 5 11.18 95.05 2.14 2 6.21 5 10.83 3 6.47 5 11.22 4 6.43 5 11.31 5 7.23 5 11.97 6 6.97 5 11.85 -
选取3批不同批号的单味药材,参照“2.1.3”的方法,制备3批当归六黄汤复方溶液,测定不同批次当归六黄汤4种主要成分的含量。同时制备各单味药材溶液,通过测定单味药材中不同成分的含量明确主要成分的来源。阿魏酸、黄柏碱、毛蕊异黄酮和巴马汀的质量分数见表3。其中,当归、黄柏和黄连中均有阿魏酸,黄柏中阿魏酸含量最高。黄柏碱仅在黄柏中检出,其他单味药中均未检出黄柏碱。黄芩和黄芪中含有毛蕊异黄酮,黄芪中含量较高。黄柏和黄芪中均有巴马汀,黄芪中巴马汀的含量较高。生地黄和熟地黄中均未检出这4种主要成分,相关文献报道地黄中的主要成分为梓醇、糖类、氨基酸和地黄素等[18-19]。生物碱、有机酸并不是地黄中的主要成分。
表 3 当归六黄汤剂和单味药材中4种成分的含量
样品(批号) 质量分数(mg/g,n=3) 阿魏酸 黄柏碱 毛蕊异黄酮 巴马汀 S1 0.878±0.008 0.447±0.006 2.897±0.044 2.310±0.047 S2 0.745±0.012 0.466±0.008 2.634±0.043 2.375±0.042 S3 0.782±0.014 0.545±0.011 3.140±0.036 2.355±0.018 当归(20210723) 0.956±0.035 / / / 生地黄(20210401-1) / / / / 熟地黄(210801) / / / / 黄芩(20210603-1) / / 22.344±0.945 / 黄柏(210803) 7.475±0.192 3.461±0.146 / 6.073±0.715 黄连(210601) 0.428±0.017 / / / 黄芪(20210318-1) / / 0.621±0.001 17.389±0.07 -
采用HPLC法进行样品的在线分离,在建立方法的过程中对色谱中的流动相及洗脱梯度进行考察。甲醇对各成分的洗脱能力较好,并能很好的改善峰形。而且在水相中加入0.5%的甲酸都能够改善生物碱成分的拖尾,在对洗脱梯度进行优化时,最终采用“2.2.1”项下的洗脱梯度,能够实现各组分之间很好的分离并获得较好的峰形。
-
黄柏碱、巴马汀、毛蕊异黄酮和阿魏酸在质谱分析中,正离子的响应较负离子的响应好,在正离子模式下,以上成分母离子存在形式为[M+H]+,通过二级裂解,选取裂解稳定响应较好的离子对进行分析。同时对多反应监测模式下质谱参数DP、CE值进行优化。其中,阿魏酸DP: 55 V,CE: 23 V;巴马汀DP: 76 V,CE: 39 V;毛蕊异黄酮DP: 75 V,CE: 33 V;黄柏碱DP: 66 V,CE: 34 V;使峰形和响应强度达到最优状态。
本文基于当归六黄汤的主要成分建立了HPLC-QTrap的定量分析检测方法,用以定量分析当归六黄汤中的阿魏酸、黄柏碱、巴马汀和毛蕊异黄酮4种主要成分的含量,该方法可用于当归六黄汤的质量控制和评价研究,也能为深入研究中药有效成分在体内的药动学研究提供方法借鉴。
Determination of four different components in Danggui Liuhuang decoction by HPLC-MS/MS
-
摘要:
目的 建立HPLC-MS/MS的分析方法同时测定当归六黄汤中4种主要成分黄柏碱、巴马汀、毛蕊异黄酮、阿魏酸的含量,为当归六黄汤的质量控制提供方法学参考和借鉴。 方法 基于HPLC-MS/MS的分析方法,质谱检测采用ESI源MRM的正离子数据采集模式。色谱柱为Agilent Extend-C18(5 μm, 4.6 mm×250 mm),甲醇和含0.5%的甲酸水溶液进行梯度洗脱。 结果 黄柏碱的线性范围为2~200 nmol/ml,巴马汀、毛蕊异黄酮、阿魏酸的线性范围均为20~2 000 nmol/ml,当归六黄汤7批样品中4种成分的含量相对稳定,其中,阿魏酸主要存在于黄柏和黄连中;黄柏碱只在黄柏中测到;毛蕊异黄酮在黄芩和黄芪中含量较高;巴马汀在黄柏和黄芪中都能检测到。 结论 该方法灵敏度高,专属性好,样品的稳定性良好,能够满足中药复方定量分析的要求,可用于当归六黄汤的质量控制和评价研究。 Abstract:Objective To establish the method of simultaneous determination of four main components of Danggui Liuhuang Decoction, including phellodendrine, palmatine, calycosin, and ferulic acid and provide reference for the quality control of Danggui Liuhuang Decoction. Methods Based on the HPLC-MS/MS analysis method, the positive ion data acquisition mode were adopted for the mass spectrometry detection and the four main components were quantified with multiple reaction monitoring mode (MRM) by ESI source. The chromatographic column was Agilent Extend-C18 (5 μm, 4.6 mm×250 mm), and gradient elution was performed with methanol and 0.5% formic acid in water. Results The linear range of phellodendrine was from 2-200 nmol/ml, and the linear range of palmatine, calycosin and ferulic acid was from 20-2 000 nmol/ml. The contents of the four components in the seven batches of Danggui Liuhuang Decoction were relatively stable, among which ferulic acid was mainly found in Phellodendrine and Coptidis; Phellodendrine was only detected in cortex phellodendri; the content of calycosin in Scutellaria baicalensis and Astragalus was higher; palmatine was detected in both Phellodendron and Astragalus. Conclusion The method had high sensitivity, good specificity and sample stability, which could meet the requirements of quantitative analysis of Traditional Chinese Medicine compounds, and could provide reference for further pharmacokinetics study on the content changes of traditional Chinese medicine compounds in biological samples. -
Key words:
- Danggui Liuhuang decoction /
- phellodendron /
- palmatine /
- calycosin /
- ferulic acid /
- content determination
-
光动力治疗(PDT)基于光辐照聚集光敏剂的肿瘤组织,由光敏剂诱发光动力反应形成单线态氧(1O2)等活性氧(ROS),通过对肿瘤细胞和肿瘤血管的直接杀伤及激活机体系统免疫反应等多种机制发挥抗肿瘤作用[1-3]。二氢卟吩及菌绿素类光敏剂是PDT新药研究的热点[4-8]。其中,已获批上市的代表药物有他拉泊芬(talaporfin)和帕利泊芬(padeliporfin)等[9, 10]。
光敏剂作为结构非特异性药物,存在缺乏肿瘤靶向性摄入和明确的作用药靶等缺陷。此外,PDT受制于局部治疗,对浸润较深的肿瘤组织,及已发生转移的肿瘤疗效有限。目前,PDT和化疗联用是克服上述缺陷,提高PDT疗效最为普遍和有效的策略之一。研究表明,抗代谢化疗药物氟尿嘧啶(5-Fu)与PDT联用具有协同抗肿瘤作用[11-13]。据此,我们设想利用在肿瘤微环境下能响应性断裂的连接基团(linker)将光敏剂与化疗药物偶联,希望实现二者在肿瘤组织的靶向释放,从而发挥其PDT和化疗协同抗肿瘤作用。酰腙键是酸敏感化学键,常被用来连接载体,以药物制备智能药物载体。这种药物载体到达肿瘤细胞的内涵体或溶酶体中时,会发生酸性水解将药物有效释放出来。因此,本文针对肿瘤微环境呈弱酸性的特点,采用药物化学最经典的前药设计策略,以脱镁叶绿素a(Phephorbide a)粗提物经酸碱降解制得的二氢卟吩e6(3)[14]为先导光敏剂,通过其152-羧基与抗肿瘤药物5-Fu以酸敏感酰腙键连接,设计合成pH响应型光化疗协同抗肿瘤光敏剂二氢卟吩e6-偕氟尿嘧啶(1),并考察其体外PDT抗肿瘤活性和pH响应性5-Fu释放,及其对黑色素瘤B16-F10和肝癌HepG2细胞的光动力抗癌活性及其作用机制,以期获得高效、低毒的PDT治癌药物候选药物,合成路线见图1。
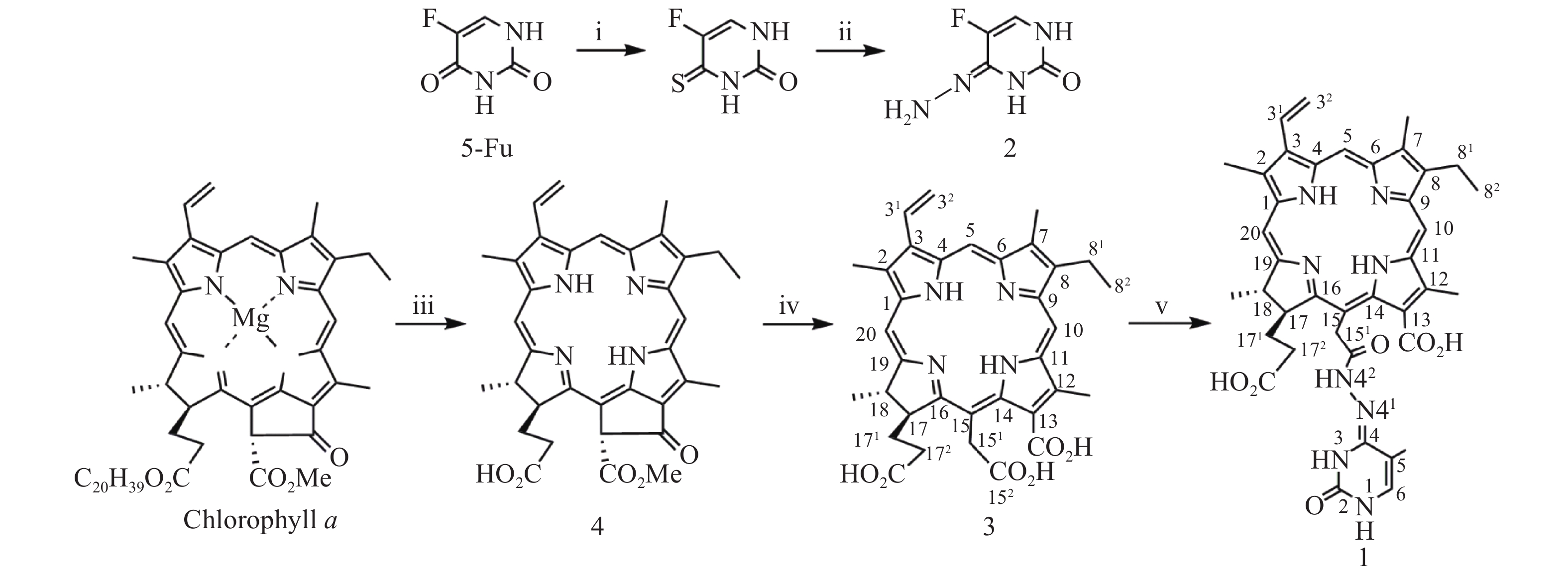 图 1 二氢卟吩e6 -偕氟尿嘧啶光敏剂(1)的合成路线试剂和反应条件:(i)五硫化二磷,吡啶,回流12 h;(ii)水合肼,甲醇,室温2 h;(iii)浓盐酸,乙醚,4 ℃ 30 min;(iv)25%氢氧化钾甲醇液,回流30 min;(v)a. EDC·HCl,N,N-二甲基甲酰胺,室温8 h;b.二异丙基乙胺,2, N,N-二甲基甲酰胺,室温12 h。
图 1 二氢卟吩e6 -偕氟尿嘧啶光敏剂(1)的合成路线试剂和反应条件:(i)五硫化二磷,吡啶,回流12 h;(ii)水合肼,甲醇,室温2 h;(iii)浓盐酸,乙醚,4 ℃ 30 min;(iv)25%氢氧化钾甲醇液,回流30 min;(v)a. EDC·HCl,N,N-二甲基甲酰胺,室温8 h;b.二异丙基乙胺,2, N,N-二甲基甲酰胺,室温12 h。1. 化学合成
1.1 仪器与试剂
用Bruker MSL-600型核磁共振仪测定1H NMR,CD3OD为溶剂;用API-3000 LC-MS型电喷雾质谱仪测定质谱(ESI-MS);用岛津UV-160型紫外分光光度计测定UV吸收谱;用日立F-7000荧光分光光度计测定荧光发射谱;用Shimazu LC-20AD HPLC仪测定化合物1的相对纯度及其5-Fu的体外释放。色谱柱型号为Waters Xterra C18柱,流动相:乙腈-0.3%乙酸水溶液(80 : 20);流速:1.0 ml/min;检测波长:400 nm(化合物1的相对纯度)或254 nm(5-Fu释放);柱温:30 ℃;进样量:20 μl。柱色谱分离用TELEDYNE ISCO的快速制备色谱Combi Flash@Rf+仪,硅胶H作为固定相。PDT抗癌活性测试使用BWT半导体激光仪(北京凯普林,波长为660 nm);用流式细胞仪(BD Accuri C6,美国)(激发波长:488 nm,发射波长:525 nm)检测受试肿瘤细胞样品的ROS水平、细胞凋亡率和细胞周期阻滞。
二氢卟吩e6(3)按照文献[14]的方法制备;其它实验用材料和化学试剂均为市售商品。
1.2 42-N-(二氢卟吩 e6-152-酰基)-5-氟尿嘧啶-4-腙(1)的合成
取氟尿嘧啶(0.2 g,1.563 mmol)溶于无水吡啶(10 ml),加入五硫化二磷(0.298 g,1.563 mmol),加热回流12 h。反应完毕,减压回收溶剂,残物加乙酸乙酯溶解(100 ml),用0.1 mol/L HCl洗涤(50 ml×2),无水Na2SO4干燥,减压除去溶剂得4-硫代-5-氟尿嘧啶粗品。上述4-硫代-5-氟尿嘧啶粗品加甲醇(10 ml)溶解,于0 ℃下滴加N2H4·H2O(0.316 g,6.252 mmol),室温继续搅拌2 h。反应完毕,减压抽滤,P2O5真空干燥得固体化合物5-氟尿嘧啶-4-腙(2)中间体,直接用于下步反应。取二氢卟吩e6(0.1 g,0.168 mmol)溶于无水DMF(10 ml),加1-乙基-(3-二甲氨基丙基)碳二亚胺盐酸盐(EDC·HCl)(0.035 g,0.183 mmol),室温搅拌反应6 h后再加入中间体2(0.031 g,0.218 mmol),继续搅拌36 h。反应完毕,反应液加入10倍体积量乙酸乙酯,饱和NaCl水溶液洗涤(50 ml×3),无水Na2SO4干燥,减压回收溶剂所得固体经快速制备色谱梯度洗脱分离纯化(流动相为二氯甲烷/甲醇/甲酸=15∶1∶0.1~8∶1∶0.1)得黑色固体1纯品0.048 g,产率39.6%。UV-vis λmax (MeOH, nm) (ε, M−1cm−1):660 (3.15×104), 510 (0.82×104), 402 (8.13×104)。1H-NMR (600 MHz, CD3OD, δ, ppm): 9.79 (s, 1H, 10-CH), 9.73 (s, 1H, 5-CH), 9.07 (s, 1H, 20-CH), 8.19 (dd, J = 18.0, 12.0 Hz, 1H, 31-CH), 7.29 (s, 1H, 5-Fu的6-CH), 6.38 (d, J = 18.0 Hz, 1H, 32-CHB), 6.15 (d, J = 12.0 Hz, 1H, 32-CHA), 5.35 (s, 2H, 151-CH2), 4.65 (m, 2H, 17-CH和18-CH), 3.84 (q, J = 7.5 Hz, 2H, 81-CH2), 3.63 (s, 3H, 12-CH3), 3.53 (s, 3H, 2-CH3), 3.30 (s, 3H, 7-CH3), 2.3~2.0 (m,4H , 171-CH2 和172-CH2), 1.76 (m, 6H¸ 18-CH3和82-CH3)。MS (ESI+) m/z: 723.63 (M+H)+ (100%)。元素分析(C38H39N8O6F,%)计算值:C 63.16, H 5.40, N 15.48;实测值:C 63.34, H 5.38, N 15.43。HPLC测定纯度:95.2%。
2. 体外光理化性质和光生物活性
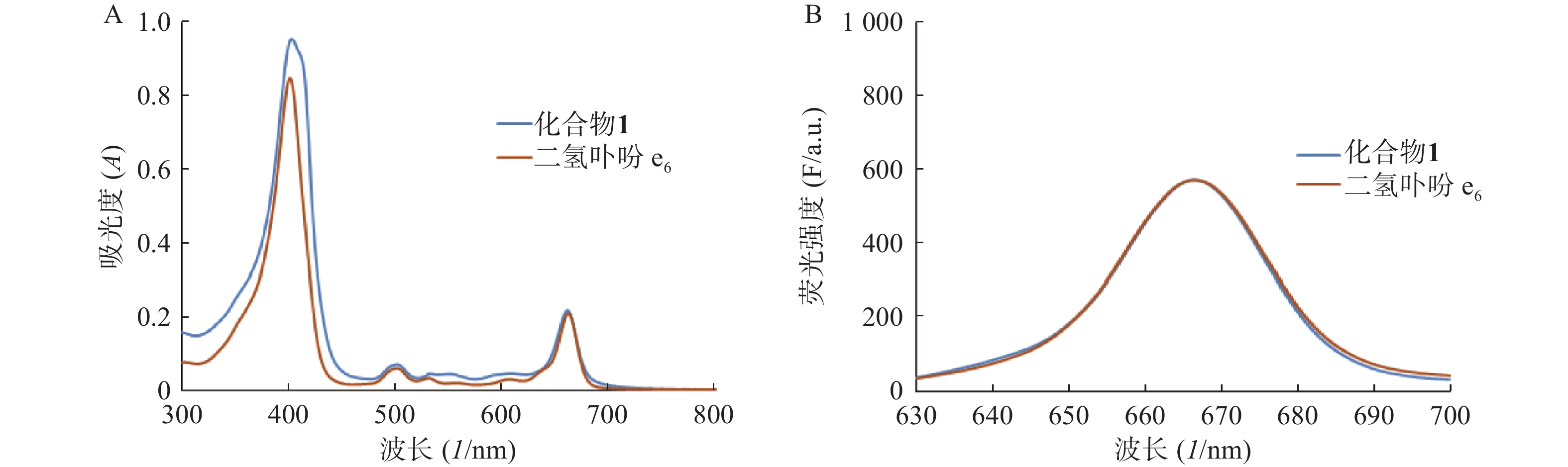
2.1 化合物1的紫外吸收谱和荧光发射谱
分别测定目标化合物1及其先导化合物二氢卟吩e6(3)的甲醇溶液(10 μmol/L)在300~800 nm处的紫外吸收谱和激发波长为400 nm的荧光发射光谱,结果见图2。
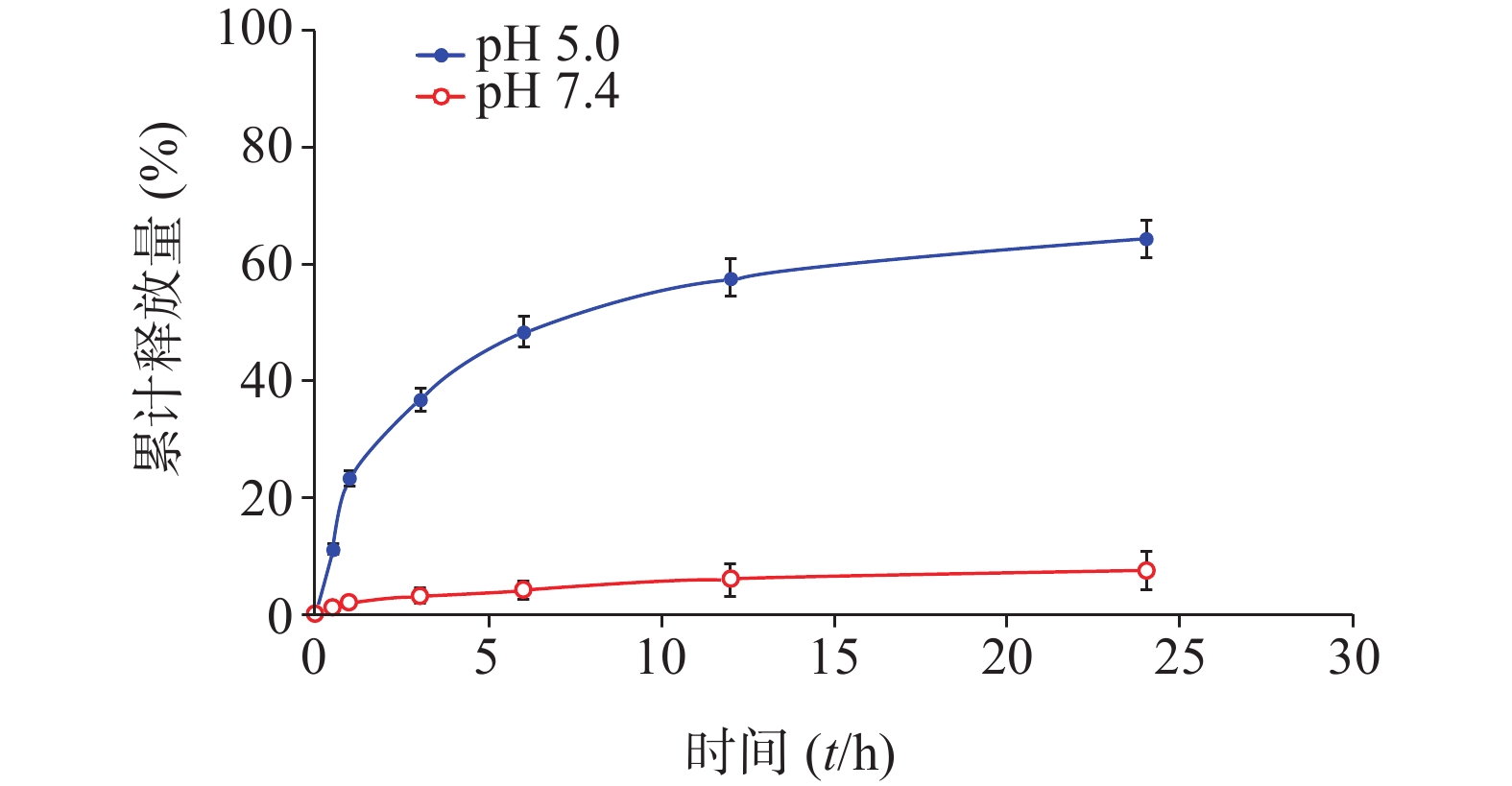
2.2 化合物1的体外pH响应性5-Fu释放
分别配制浓度为50 μmol/L的化合物1的HOAc-NaOAc缓冲液(pH 5.0)和PBS溶液(10 ml),并于0.5、1.0、3.0、6.0、12、24 h时分别取样(500 μl)。其中,HOAc-NaOAc缓冲液(pH 5.0)组取样液用0.1 mol/L氢氧化钠水溶液迅速调节pH值至7.4。每份取样液加PBS稀释至原溶液1/3浓度,微孔滤膜(孔径0.22 μm)过滤,HPLC进样检测;实验重复3次。根据5-Fu的HPLC峰面积-浓度标准曲线分析计算,绘制目标化合物1于弱酸(pH 5.0)中的5-Fu体外释放量-时间曲线,结果见图3。
2.3 化合物1的体外光动力抗癌活性
2.3.1 细胞孵育
2.3.2 细胞暗毒性测试
参照文献[6-8]的方法,每孔5×103个B16-F10细胞或HepG2细胞悬液(100 μl)接种于96孔板上,加入等体积上述细胞培养液孵育24 h;更换含不同浓度待测物的培养液(DMSO浓度小于1%,100 μl),继续避光孵育48 h;再更换含10%(V/V)CCK-8(Beyotime,中国)的RPMI 1640基础培养基(100 μl),继续培养1.5 h,然后用Varioskan Flash全波长酶标仪(Thermo)于波长450 nm处测定每孔的吸光度值,计算各浓度对应的细胞存活率,并拟合得到待测物的肿瘤细胞半数抑制浓度即IC50值。
2.3.3 细胞光毒性测试
每孔5×103个B16-F10细胞或HepG2细胞悬液(100 μl)接种于96孔板上,加入等体积细胞培养液孵育24 h;更换含不同浓度待测物的细胞培养液(DMSO浓度小于1%,100 μl),继续避光孵育24 h;再更换新鲜培养液(100 μl),以波长为660 nm的激光辐照受试细胞样品(光照剂量为10 J/cm2),继续孵育24 h。最后按“2.3.2”项下CCK-8法测定各待测物的肿瘤细胞IC50值。
2.3.4 实验结果
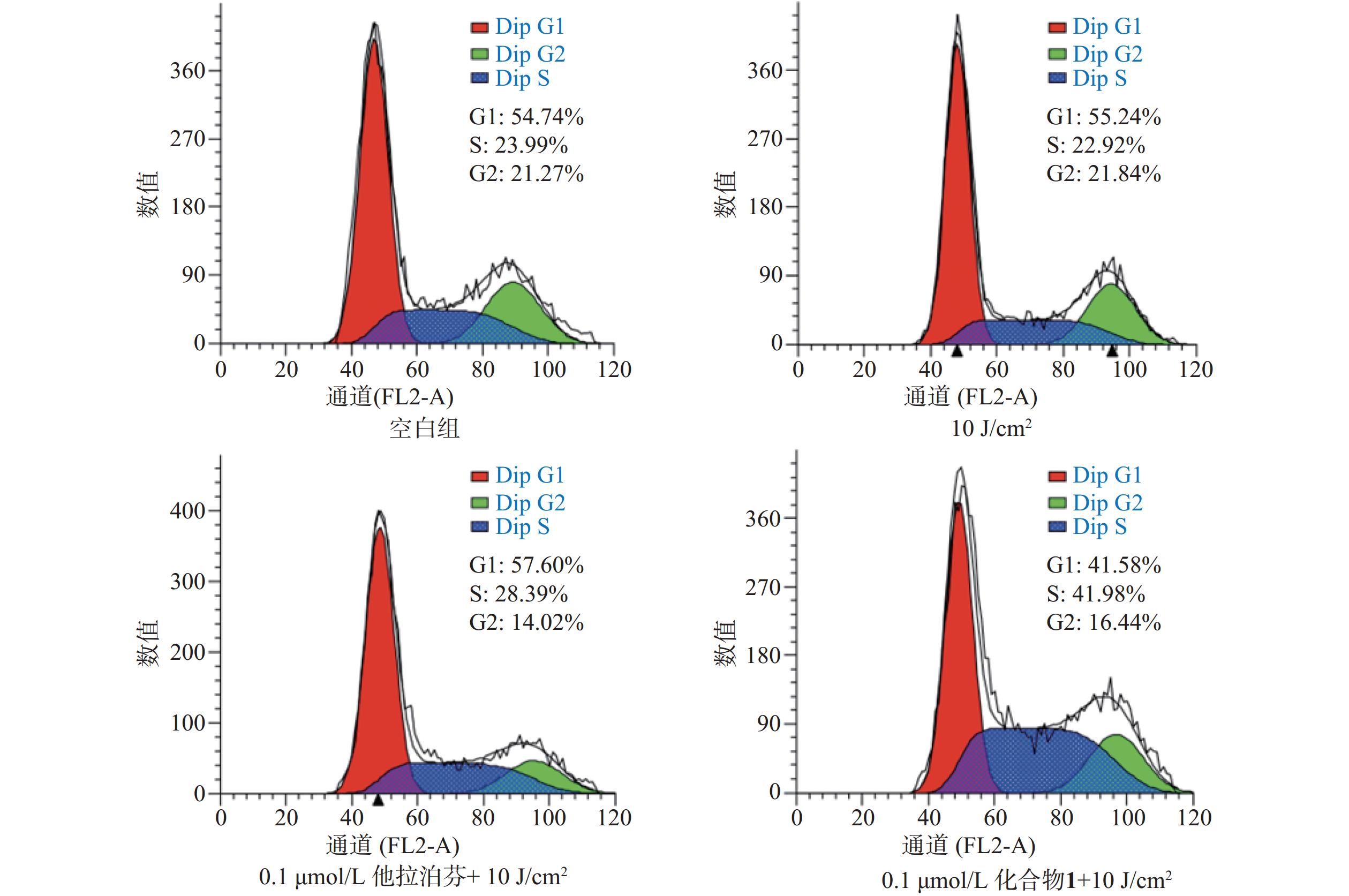
以临床光敏药物他拉泊芬为阳性对照,化合物1及其先导化合物3对肿瘤细胞株的体外PDT抗癌活性结果见表1。
表 1 目标化合物1的体外光动力抗癌活性(IC50,μmol/L)化合物 B16-F10细胞 暗毒/光毒比 HepG2细胞 暗毒/光毒比 暗毒性 光毒性 暗毒性 光毒性 化合物 1 46.84±8.46*, ΔΔΔ 0.73±0.16**, ΔΔΔ 64.2 50.80±6.45**, #, ΔΔΔ 0.90±0.22**, ΔΔΔ 56.4 二氢卟吩e6 69.72±4.69 3.36±0.59 20.8 70.38±10.9 2.75±0.41 25.6 他拉泊芬 254.8±18.8 11.31±3.88 22.5 176.4±28.4 15.47±5.07 11.4 5-Fu 35.80±6.68 NTa − 39.16±2.7 NTa − NTa:未测定;*P < 0.05,**P < 0.01,与二氢卟吩 e6组比较;#P < 0.05,与5-Fu组比较;ΔΔΔP < 0.001,与他拉泊芬组比较。 2.4 化合物1介导的PDT对肿瘤细胞内ROS水平的影响
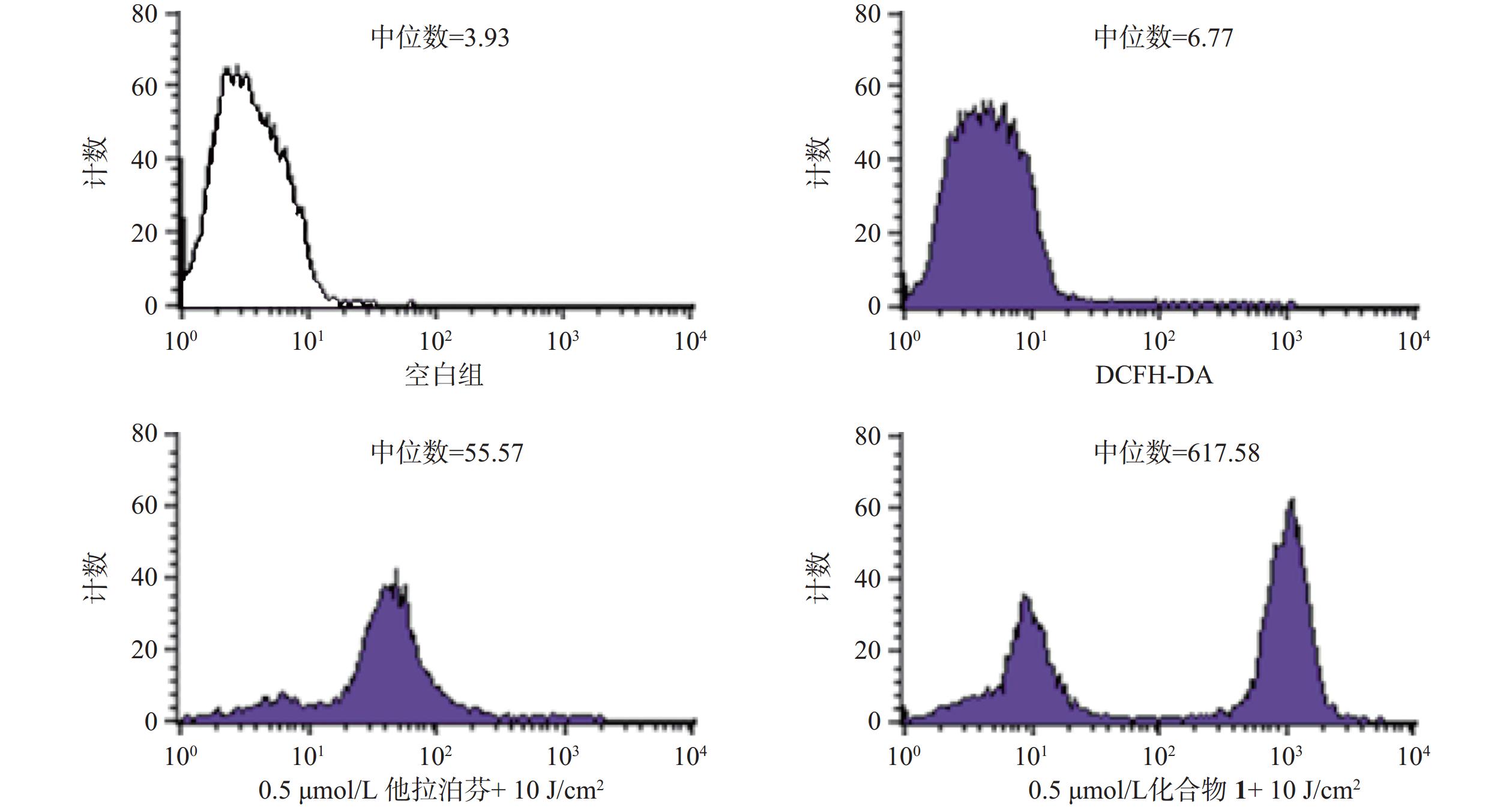
操作步骤如下:a. 每孔3 × 105个B16-F10细胞悬液(2 ml)接种6孔板上,按“2.3.1”项条件避光孵育24 h;b. 分别更换含一定浓度化合物1或他拉泊芬的新鲜培养液(DMSO浓度小于1%,2 ml),继续避光孵育24 h;c. 加入10 mmol/L DCFH-DAROS荧光检测探针(Beyotime,1.5 μl),吹打混匀,继续避光孵育20 min;d. PBS洗涤3次,再加新鲜培养液(2 ml),以660 nm波长的激光辐照(光剂量10 J/cm2)细胞样品,继续避光孵育20 min;e. 收集每孔细胞样品,用流式细胞仪检测各孔细胞ROS水平,结果见图4。
2.5 化合物1介导的PDT对肿瘤细胞凋亡的影响
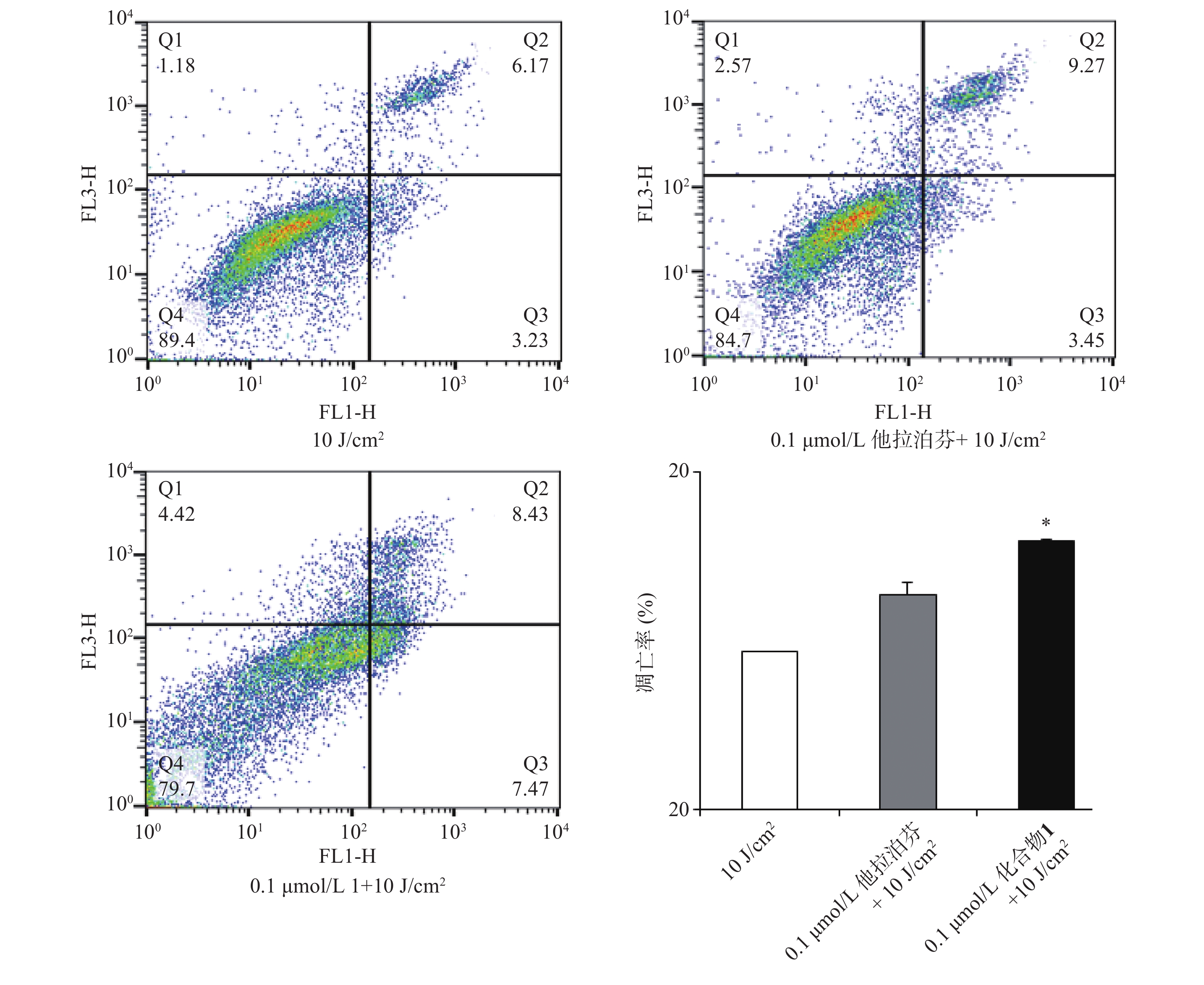
按“2.4”项下操作方法,仅从步骤c开始,更换新鲜培养液(2 ml),用660 nm波长的激光辐照(光剂量10 J/cm2)细胞样品,继续避光孵育20 min;d. 以1 500 r/min离心(5 min)细胞样品,PBS洗涤,再以1 000 r/min离心(5 min)后获取细胞样品;e. 按Annexin V-FITC细胞凋亡检测试剂盒(Beyotime)操作流程操作,结果见图5。
2.6 化合物1介导的PDT对肿瘤细胞周期的阻滞作用
按“2.5”项下操作方法,仅在e步骤中,换以细胞周期阻滞检测试剂盒(Beyotime)的操作流程,每份细胞样品中分别加入染色缓冲液(300 µl)、RNase A(6 µl)和碘化丙啶染色液(15 µl),轻轻混匀,避光孵育20 min后,用流式细胞仪进行细胞周期阻滞检测,结果见图6。
3. 结果与讨论
按文献[14]方法制得的二氢卟吩e6(3)为先导化合物,经1-乙基-(3-二甲氨基丙基)碳二亚胺盐酸盐(EDC·HCl)于无水DMF中催化分子内脱水缩合制得二氢卟吩e6-131,152-酸酐活泼中间体[15],然后直接与中间体2发生酰化反应成功合成得到了光化疗双模抗肿瘤光敏剂二氢卟吩e6-偕氟尿嘧啶(1),反应收率达39.6%,其结构经UV、ESI-MS、1H NMR及元素分析确证。
化合物1在甲醇中最大紫外吸收波长和荧光发射波长(激发波长:400 nm)分别为660 nm和670 nm,与先导物3相一致,表明先导物3以酰腙键偶联5-Fu后,并没有改变其作为光敏剂特有的紫外吸收和荧光发射光谱等光物理特性。此外,化合物1在弱酸(pH 5.0)条件下,能有效释放5-Fu,24 h内累积释放率可达60.3%;但在pH 7.4的条件下较为稳定,24 h内5-Fu累积释放率仅为5%。
体外PDT抗癌活性测试结果显示,化合物1对B16-F10和HepG2细胞株的光毒活性和暗毒/光毒比(治疗指数)均显著优于先导物二氢卟吩e6(3)(P<0.005)和他拉卟吩(P<0.001),其IC50值分别达0.73 μmol/L和0.90 μmol/L。
体外PDT抗癌机制研究提示,化合物1介导的PDT能显著提升B16-F10细胞内ROS水平和诱导B16-F10细胞凋亡,并阻滞肿瘤细胞周期于S期。
总之,二氢卟吩e6-偕氟尿嘧啶(1)具有PDT抗癌活性强、治疗指数(暗毒/光毒比)高且可在肿瘤弱酸环境中有效释放5-Fu等优点,从而实现“单分子”光化疗协同抗肿瘤作用,值得进一步开发研究。
-
图 1 不同样品的提取离子流色谱图
A.20 nmol/ml混合对照品提取总离子流色谱图;B.当归六黄汤复方供试品溶液提取总离子流色谱图;C.黄柏碱提取离子流色谱图;D.巴马汀的提取离子流色谱图;E.阿魏酸提取离子流色谱图;F.毛蕊异黄酮提取离子流色谱图1.黄柏碱;2.巴马汀;3.阿魏酸;4.毛蕊异黄酮。
表 1 4种成分的标准曲线方程、相关系数、线性范围
成分 标准曲线方程 相关系数r 浓度范围
(nmol/ml)阿魏酸 Y=443X+73.8 0.999 2 20~2 000 黄柏碱 Y=4.76×104X+1.57×104 0.995 8 2~200 巴马汀 Y=1.02×104X−4.35×103 0.996 7 20~2 000 毛蕊异黄酮 Y=1.54×103X+73.8 0.999 6 20~2 000  下载: 导出CSV
下载: 导出CSV
表 2 4种成分的提取回收率(n=6)
成分 序号 已知浓度
(nmol/ml)加入浓度
(nmol/ml)测得浓度
(nmol/ml)提取
回收率
(%)RSD
(%)阿魏酸 1 22.50 25 46.82 97.74 3.09 2 24.17 25 50.06 3 18.10 25 42.54 4 17.83 25 41.33 5 23.63 25 48.26 6 19.84 25 43.67 巴马汀 1 32.81 30 63.57 98.09 2.87 2 36.29 30 65.61 3 32.74 30 61.52 4 34.18 30 63.13 5 33.42 30 61.83 6 30.92 30 61.25 毛蕊异黄酮 1 50.82 50 101.74 102.50 1.20 2 53.50 50 103.86 3 51.22 50 103.12 4 49.31 50 100.53 5 51.08 50 103.24 6 55.33 50 106.26 黄柏碱 1 6.54 5 11.18 95.05 2.14 2 6.21 5 10.83 3 6.47 5 11.22 4 6.43 5 11.31 5 7.23 5 11.97 6 6.97 5 11.85
下载: 导出CSV
表 3 当归六黄汤剂和单味药材中4种成分的含量
样品(批号) 质量分数(mg/g,n=3) 阿魏酸 黄柏碱 毛蕊异黄酮 巴马汀 S1 0.878±0.008 0.447±0.006 2.897±0.044 2.310±0.047 S2 0.745±0.012 0.466±0.008 2.634±0.043 2.375±0.042 S3 0.782±0.014 0.545±0.011 3.140±0.036 2.355±0.018 当归(20210723) 0.956±0.035 / / / 生地黄(20210401-1) / / / / 熟地黄(210801) / / / / 黄芩(20210603-1) / / 22.344±0.945 / 黄柏(210803) 7.475±0.192 3.461±0.146 / 6.073±0.715 黄连(210601) 0.428±0.017 / / / 黄芪(20210318-1) / / 0.621±0.001 17.389±0.07
下载: 导出CSV
-
[1] 杨玲, 彭江丽, 李娟, 等. 当归六黄汤的药理作用和临床应用研究进展[J]. 中国实验方剂学杂志, 2021, 27(2):233-241. [2] CAO H, TUO L L, TUO Y L, et al. Immune and metabolic regulation mechanism of Dangguiliuhuang Decoction against insulin resistance and hepatic steatosis[J]. Front Pharmacol, 2017, 8:445. doi: 10.3389/fphar.2017.00445 [3] 吴青华, 李冰涛, 涂珺. 复方中药治疗糖尿病的研究进展[J]. 中国中药杂志, 2019, 44(6):1104-1109. [4] 李志悦, 刘香春, 蒲蔚荣, 等. 当归六黄汤加减方治疗阴虚火旺型甲亢疗效观察[J]. 陕西中医, 2017, 38(7):914-915. [5] 杨明, 张定堃, 钟凌云, 等. 对传统中药炮制文化与哲学的思考[J]. 中国中药杂志, 2013, 38(13):2223-2226. [6] 李海英, 贺鹏, 贺玉婷, 等. 中药复方配伍研究的关键问题及超分子化学解决对策[J]. 中草药, 2019, 50(12):2757-2762. [7] LIU T T, CAO H, JI Y C, et al. Interaction of dendritic cells and T lymphocytes for the therapeutic effect of Dangguiliuhuang Decoction to autoimmune diabetes[J]. Sci Rep, 2015, 5:13982. doi: 10.1038/srep13982 [8] 邱敏, 邹文娟, 陶劲, 等. 当归六黄汤主治病机探微[J]. 中国中医基础医学杂志, 2017, 23(12):1665-1666. [9] 张永. 基于医案数据分析的当归六黄汤组方与临床应用研究[D]. 成都: 成都中医药大学, 2018. [10] 计雅纯. 中药复方当归六黄汤质量控制研究和XEDJ药效学研究[D]. 武汉: 华中科技大学, 2016. [11] 黄明军, 孙耀志, 高松, 等. LC-MS分析当归六黄汤中主要成分[J]. 中国实验方剂学杂志, 2016, 22(9):63-67. [12] 闫妍, 宿莹, 武艳雪, 等. 当归六黄汤本草考证及物质基准的制备工艺研究[J]. 医药导报, 2021, 40(10):1403-1407. [13] 张留记, 王建霞, 屠万倩, 等. 生地黄与熟地黄中5个苷类成分和总多糖的含量比较[J]. 天然产物研究与开发, 2019, 31(4):566-571. [14] 严斐霞, 谢永艳, 陈畅, 等. 熟地黄炮制过程中的化学成分变化和药理作用研究进展[J]. 时珍国医国药, 2021, 32(10):2493-2495. [15] 董开心, 许军, 刘燕华, 等. 新型双酯类阿魏酸衍生物的合成及降血脂活性研究[J]. 中国药物化学杂志, 2022, 32(2):83-89. [16] 李修刚, 张玲钰. 阿魏酸的合成与应用研究进展[J]. 山东化工, 2020, 49(17):81-82. [17] 于玲, 王知斌, 王秋红, 等. 黄芪中黄酮类化合物药理作用研究进展[J]. 中医药信息, 2018, 35(2):104-108. [18] 龚小保. 黄连生物碱改善糖尿病周围神经病变活性成分筛选及作用机制研究[D]. 重庆: 西南大学, 2022. [19] 徐文慧, 常丽静, 段连政, 等. 黄芪不同部位黄芪甲苷及毛蕊异黄酮葡萄糖苷的含量测定[J]. 吉林中医药, 2020, 40(2):255-258. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5496
- HTML全文浏览量: 2561
- PDF下载量: 40
- 被引次数: 0
