

 下载:
下载:

-
据估计,2017 年全球有 4.51 亿(18~99 岁)糖尿病(DM)患者,预计到 2045 年,这一数字将增至 6.93 亿[1]。糖尿病肾病(DKD)是DM的一种严重并发症,是世界范围内终末期肾病(ESRD)的主要病因[2, 3]。DKD的发生率与DM的发病率和病死率增加密切相关[4]。在美国,开始接受ESRD治疗的DM患者数量从2000年的4万多人显著增加到2014年的5万多人[5]。在我国,DKD的发病率在过去10年中显著增加,2013年的一项调查结果显示,中国DKD患者数量估计达到2 430万[6]。当前,DM患病率持续上升,如果DKD的临床预防策略没有改善,预计DKD的患病率也会随之增加[7, 8]。然而目前除了控制血糖、血压等手段外,临床上尚无其他有效预防DKD的方案。DKD的发病机制复杂,其分子机制还没有得到全面阐明。近年来,越来越多的研究发现,肠道菌群在DKD的发生发展中发挥了重要作用,本文围绕DKD的肠道菌群参与情况,综述其研究进展。
肠道菌群是一个由微生物菌落组成的复杂生态系统,包括至少1 000个不同物种的数万亿细菌,另外还有其他共生生物,如古细菌、病毒、真菌和原生生物。肠道菌群失调的主要特征是细菌和真菌的多样性和丰度下降[9]。近年来,人们对肠道菌群与宿主相互作用产生极大兴趣,众多证据表明,肠道菌群在人类健康和疾病中发挥重要作用,菌群失调已被证明与动脉粥样硬化、高血压、心力衰竭、慢性肾病(CKD)、肥胖和2型糖尿病(T2DM)等疾病有关[10]。肠道菌群有能力产生一系列代谢产物,包括短链脂肪酸(SCFAs)、N-氧化三甲胺(TMAO)、胆汁酸(BA)、蛋白质结合的尿毒症毒素(PBUT)、支链氨基酸(BCAAs)和一些其他未知代谢产物。肠道微生物产生的代谢产物被认为是微生物与宿主之间交流的媒介,对人体的生物活性和代谢有重要影响[11]。近年来,许多研究调查了DM、肥胖和代谢综合征等代谢性疾病患者肠道微生物群的多样性和功能的变化。有研究发现,这些患者的肠道微生物群落发生了显著变化,并导致肠道微生物群失调和肠漏综合征,肠道屏障功能障碍,肠道通透性增加[12]。多种肠道微生物群代谢产物被释放到血液中,如SCFAs、TMAO、脂多糖(LPS)和尿毒症毒素,再通过多种信号通路进一步导致疾病表型的变化[13, 14]。
-
DKD是DM患者的主要微血管并发症,尽管DKD发病机制相当复杂,但最近的研究表明,肠道菌群也参与了DKD的进展。一项研究通过16s rDNA测序分析了DKD患者和健康志愿者之间粪便样本,发现与健康志愿者相比,DKD患者肠道细菌丰富度和多样性显著下降,而许多常见病原体在DKD和DM患者中均富集,如拟杆菌门、毛梭菌门、双歧杆菌门、乳杆菌门、罗斯氏菌门和粪杆菌门。其中,巨球菌属、厌氧菌属和嗜血菌属等属在DKD中的丰富度远高于DM中的丰富度。几个特征属,如巨球菌属、韦荣球菌属、埃希氏菌属、志贺氏菌属、厌氧菌属和嗜血杆菌属则可能是DKD新的潜在微生物标志物[15]。有研究发现,中国人和欧洲女性DM患者中的哈氏梭菌增加,而罗斯伯里氏菌减少,研究者推测这与DM的发展有关[16, 17]。另一项研究则把DKD患者与DM患者进行比较,发现与DM患者相比,DKD患者肠道疣微菌门和梭杆菌门的水平显著升高,二者都属于G-菌,而LPS可通过加速巨噬细胞/单核细胞和中性粒细胞的活化而诱发炎症,导致DKD进展[18]。CKD患者常出现肠道菌群失调、细菌代谢产物累积、肠道屏障功能破坏和慢性炎症,大多数CKD患者肠道细菌过度生长,但细菌多样性下降,如梭杆菌属等在ESRD 患者体内显著富集,而产生SCFAs的细菌,尤其是产生丁酸的细菌丰度随着 ESRD 的恶性进展而逐渐下降,SCFAs通常被认为在维持人类健康方面发挥着多种重要作用[19, 20]。
以上研究表明,健康的肾脏通过细胞和分子信号与肠道微生物群沟通,以确保肠道微生物群的正常稳态。肠道菌群的失衡将导致这种平衡被破坏,肠道菌群多样性下降、特定菌群种类变化可能在DKD的发展中发挥重要作用。
-
肠道的黏液层和上皮结构是肠道屏障最重要的结构,肠道上皮是有正常上皮细胞和几种具有特定功能的细胞组成,包括潘氏细胞、杯状细胞等[21]。潘氏细胞可分泌溶菌酶和防御素等抗菌肽,防止有害细菌定植,杯状细胞通过分泌黏蛋白来维持黏膜层。上皮细胞通过顶端连接复合体连接,该复合体由紧密连接(TJ)和黏附连接组成。TJ由TJ蛋白Claudin、Occludin和连接黏附蛋白分子 A (JAM-A) 以及细胞内斑块蛋白组成[22]。肠道屏障将肠腔中的微生物与内部环境分隔开来,许多微生物系统通过肠壁与内部环境相互作用。DKD患者肠道微生物群组成和功能的改变会导致肠上皮屏障受损和肠道通透性增加[23]。一方面,由于DKD患者肾脏滤过减少,大量尿素水解导致氨重吸收增加,随后肝脏重新合成尿素,导致跨上皮电阻(TER)显著下降,关键TJ蛋白如Claudin-1、Occludin 和ZO-1丢失[24]。另一方面,DKD肠道菌群失调引起相应代谢产物发生变化。肠道菌群代谢会产生多种类型的SCFAs。Nathan等在一项小鼠骨髓移植研究中发现,丁酸盐通过为肠上皮细胞提供能量来维持结肠细胞健康,这可能有助于肠上皮的完整性[25]。Huang等发现,丁酸盐还可能通过改变肠道Claudin-2水平来抑制细胞因子诱导的屏障功能障碍[26]。此外,一些动物研究表明,乙酸盐可直接激活核苷酸结合寡聚物肠上皮细胞中的炎症结构域3(NLRP3)炎症小体,导致IL-18释放,进而通过激活小鼠上皮细胞上的IL-18受体来促进肠屏障的完整性[27]。丙酸盐还可以抑制小鼠结肠组织中 ZO-1、Occludin 和上皮钙黏蛋白(E-cadherin) 下调改善由葡聚糖硫酸钠(DSS)引起的高肠通透性[28]。体外实验中,乙酸盐、丙酸盐和丁酸盐已被证明可以通过改变肠道上皮TJ蛋白的表达促进细胞内通透性,包括体外的 ZO-1[29]。综上所述,SFCAs 被认为是维持肠道屏障的关键因素。
-
BA由肝细胞中的胆固醇在肝脏中合成。初级BA可以通过肠道微生物群转化并分解为次级 BA。肠道微生物群通过初级BA的解结合、脱氢和二羟基化来调节 BA 代谢过程[30]。BA 是G蛋白偶联胆汁酸受体 (TGR5)和核激素受体法尼醇X受体(FXR)的配体。BA与TGR5结合,通过胰高血糖素样肽-1 (GLP-1)提高胰岛素敏感性,并调节肌肉或棕色脂肪组织的能量消耗[31]。FXR的激活会减少脂肪生成和肝脏糖异生,并通过产生抗菌肽来抑制细菌过度生长和易位[32]。Wang等在链脲霉素诱导小鼠糖尿病实验中发现, FXR和TGR5通过调节肾脏信号通路在DM和肥胖相关肾脏疾病中发挥肾脏保护作用[33]。另一项体外实验证明,龙胆苦苷通过 TGR5 激活抑制 NF-κB 信号通路,从而减轻DKD中的炎症和纤维化[34]。熊去氧胆酸(UDCA)是一种继发性BA,已被发现可减轻DKD大鼠肾内质网应激引起的肾功能障碍、足细胞凋亡和氧化应激。给予牛磺酸脱氧胆酸(TUDCA)可以减轻 DM大鼠的肾小球和肾小管损伤,这部分是通过抑制内质网介导的[35]。
-
SCFAs 是肠道菌群代谢的主要产物之一,主要包括由拟杆菌门产生的乙酸盐、丙酸盐和厚壁菌门产生的丁酸盐。丁酸盐通过介导miR7a-5p/P311/TGF-β1通路缓解DKD进展, 转化生长因子-β1(TGF-β1)是触发纤维化信号级联的初始因子[36]。丁酸钠(NaB)是一种已知的核因子E2相关因子2(NRF2)激活剂,具有预防DKD的作用。研究发现,未接受NaB治疗的DM小鼠表现出明显的肾脏病理变化,如氧化损伤、炎症、细胞凋亡、纤维化。NaB 通过激活NRF2抑制组蛋白去乙酰化酶(HDAC)活性改善DKD[37]。Gasdermin D (GSDMD)是一种新发现的焦亡关键执行蛋白,可被炎症性半胱氨酸天冬氨酸酶(caspase)裂解。高糖可增加碘化丙啶(PI)阳性细胞水平,促进乳酸脱氢酶(LDH)、IL-1β、IL-18的释放,并伴有caspase-1水平升高。NaB通过NF-κB/IκB-α信号通路的caspase 1-GSDMD 经典焦烧死亡途径改善了高葡萄糖诱导的肾小球内皮细胞焦亡[37-39]。由以上研究可见,丁酸盐在高糖刺激的肾损伤中发挥重要作用,并可能作为有效的治疗靶点。此外,SCFAs改变导致肠上皮TJ的破坏,进而引起肠道通透性增加,肠腔内微生物代谢物及其他有害物质穿过肠道屏障进入体内循环,如甲酚、吲哚分子、LPS等。LPS会持续浸润门静脉,导致代谢性内毒素血症和炎症细胞因子水平升高,从而加速DKD的进展[40]。 LPS 是G-菌的表面抗原,通过 TLR2 和 TLR4 相关途径介导宿主炎症[41]。TLR2和TLR4通过诱导肿瘤坏死因子-α(TNF-α)、IL-1和IL-1β等促炎细胞因子的释放, NF-κB介导的炎症级联反应,参与DKD的持续炎症反应过程[42]。
其他SCFAs在DKD中的作用仍存在争议,在一项微生物移植治疗DM大鼠的研究中发现,乙酸盐可能会加剧 DKD的疾病进展, 如肠道微生物群产生过量的乙酸盐,通过激活G蛋白偶联受体43(GPR43)破坏胆固醇稳态,导致肾脏出现肾小管间质损伤[43]。另一项动物研究发现,肠道微生物群失调可能与早期DKD肾内肾素-血管紧张素系统(RAS)激活有关,血浆醋酸盐水平与肾内血管紧张素Ⅱ蛋白表达呈正相关,推测醋酸盐也可能参与早期DKD的肾损伤[44]。
-
TMAO主要来源于肠道菌群氧化三甲胺(TMA)。肠道微生物从摄入的卵磷脂和胆碱等营养物质中代谢并产生TMA,这些营养物质通过门静脉循环进入肝脏,并被黄素单加氧酶3或其他黄素单加氧酶氧化,产生TMAO。TMAO水平升高与CKD患者死亡风险增加相关[45]。TMAO及其前体胆碱水平的增加与信号转导蛋白SMAD3和TGF-β信号的磷酸化增强有关,从而加重高脂饮食(HFD)喂养小鼠的肾胶原沉积和肾小管间质纤维化。饮食诱导的肥胖小鼠模型中,TMAO水平升高还与NADPH氧化酶和炎症细胞因子的升高有关,而补充 TMA 形成抑制剂可改善 HFD 诱导的肾损伤纤维化并降低小鼠肾损伤分子-1(KIM-1)和促炎细胞因子的表达[46]。这些研究表明,高水平的TMAO可能是CKD进展的致病介质。近年来,研究发现TMAO在DKD发展中也发挥重要作用。TMAO可激活DKD患者的NF-κB通路,进一步加重体内微炎症,导致DKD恶化[47]。与无肾脏疾病的 T2DM 患者以及健康个体相比,DKD患者表现出更高浓度的TMAO,这与尿白蛋白肌酐比值(UACR) 呈正相关[48]。在一项动物研究中,喂食TMAO的DKD大鼠表现出更严重的肾功能衰退和肾纤维化,该研究证明TMAO可以通过激活NLRP3炎症小体并最终导致IL-1β和IL-18的释放来加速肾脏炎症[49]。
由此可见,肠道微生物群衍生的代谢物,是肠道菌群影响DKD进展的重要参与者,肠道微生物及其代谢物与肾脏之间存在相互作用,称为肠肾轴。
-
肠道微生物群与肾脏疾病相关,已有众多研究证实了肠肾轴的存在。肠道微生物群通过产生无数代谢物来参与宿主体内平衡,这些代谢物充当代谢反应的关键信号分子和底物。基于肠道微生物群的治疗可能是未来预防和治疗DKD的一种有前景的策略。饮食结构的改变有助于改善肠道菌群失调,肠道菌群移植也有望在安全、规范的条件下发挥恢复DKD微生物群生态的作用,宏基因组学和代谢组学的结合有助于研究肠道菌群失调与代谢紊乱之间的关系[49]。然而,目前大部分数据仅限于动物模型,需要更可靠的临床试验来阐明DKD发病机制的关键途径和特定菌株。
Research progress on the mechanism of gut microbiota participating in diabetes nephropathy
-
摘要: 随着糖尿病患病率升高,糖尿病肾病的预防和治疗已成为世界性难题。糖尿病肾病发生发展的分子机制目前尚不明确,但近年来诸多研究表明,肠道菌群在糖尿病肾病的进展中发挥重要作用。综述了肠道菌群参与糖尿病肾病的机制研究进展。Abstract: With the increasing prevalence of diabetes, the prevention and treatment of diabetes nephropathy have become a worldwide problem. The molecular mechanism of the occurrence and development of diabetes nephropathy is still unclear, but many studies in recent years have shown that gut microbiota plays an important role in the progress on diabetes nephropathy. The research progress on the mechanism of gut microbiota participating in diabetes nephropathy was reviewed in this article.
-
Key words:
- diabetes /
- diabetes nephropathy /
- intestinal flora /
- intestinal permeability
-
中药挥发性成分(VOCs)是指中药中一类具有芳香气并易挥发的成分,其化学组成复杂,主要包括挥发油类以及其他分子量较小、易挥发的化合物,例如萜类、脂肪族、芳香族化合物等。VOCs具有发汗解表、芳香开窍、镇咳祛痰、理气、驱风、抑菌、镇痛、杀虫等多种功效。作为中药学研究的热点之一,高效率、高标准的检测药材中VOCs十分关键。因此,利用现代化方法来实现对中草药挥发性成分的细致分析,意义巨大。
VOCs常用气相色谱-质谱联用(GC-MS)方法进行分析,虽分离能力强,但样品需要预处理且分析时间长。气相色谱-离子迁移谱(GC-IMS)法结合了GC突出的分离能力和IMS快速响应、高灵敏度的特点,具有样品准备简便、高灵敏度、高分辨率等显著优势,结合化学计量学分析中药材VOCs所呈现图谱,可实现对药材VOCs无损、快速区分[1]。
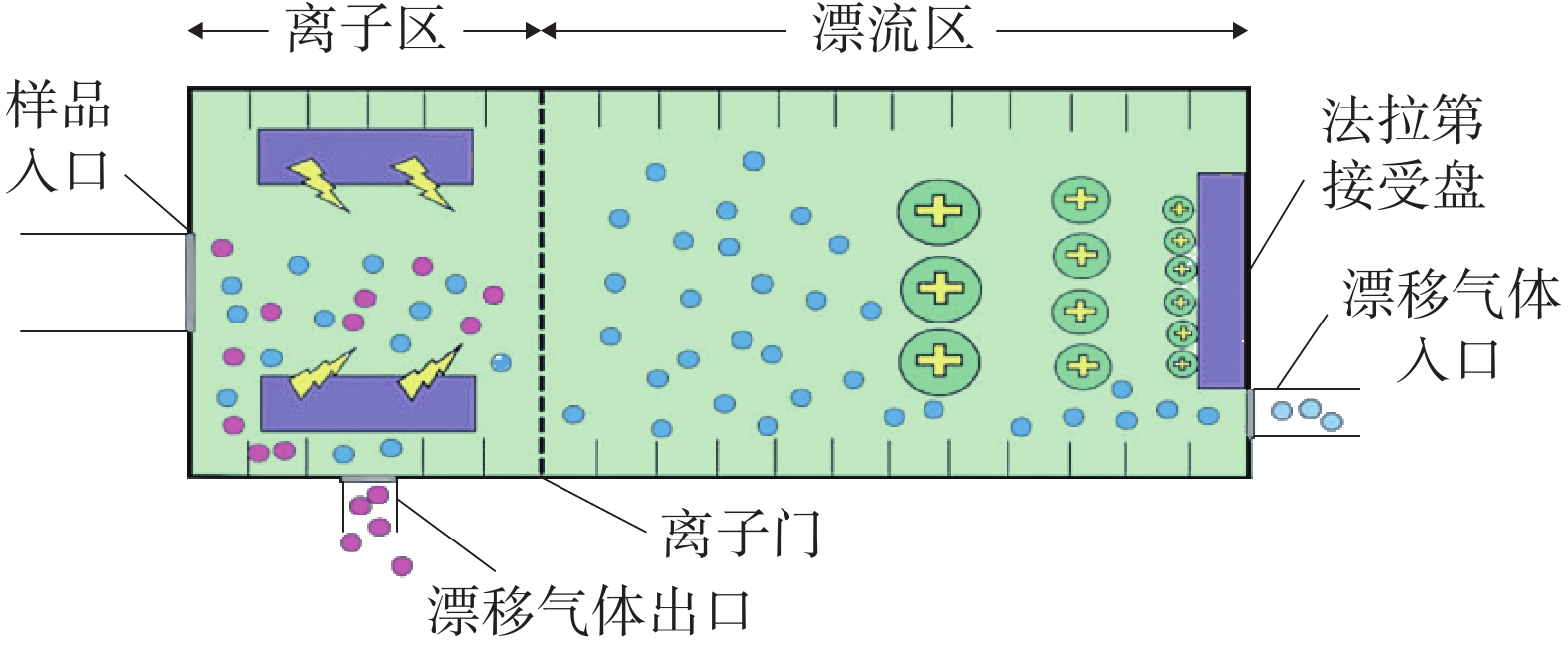
1. 气相色谱–离子迁移谱联用技术原理
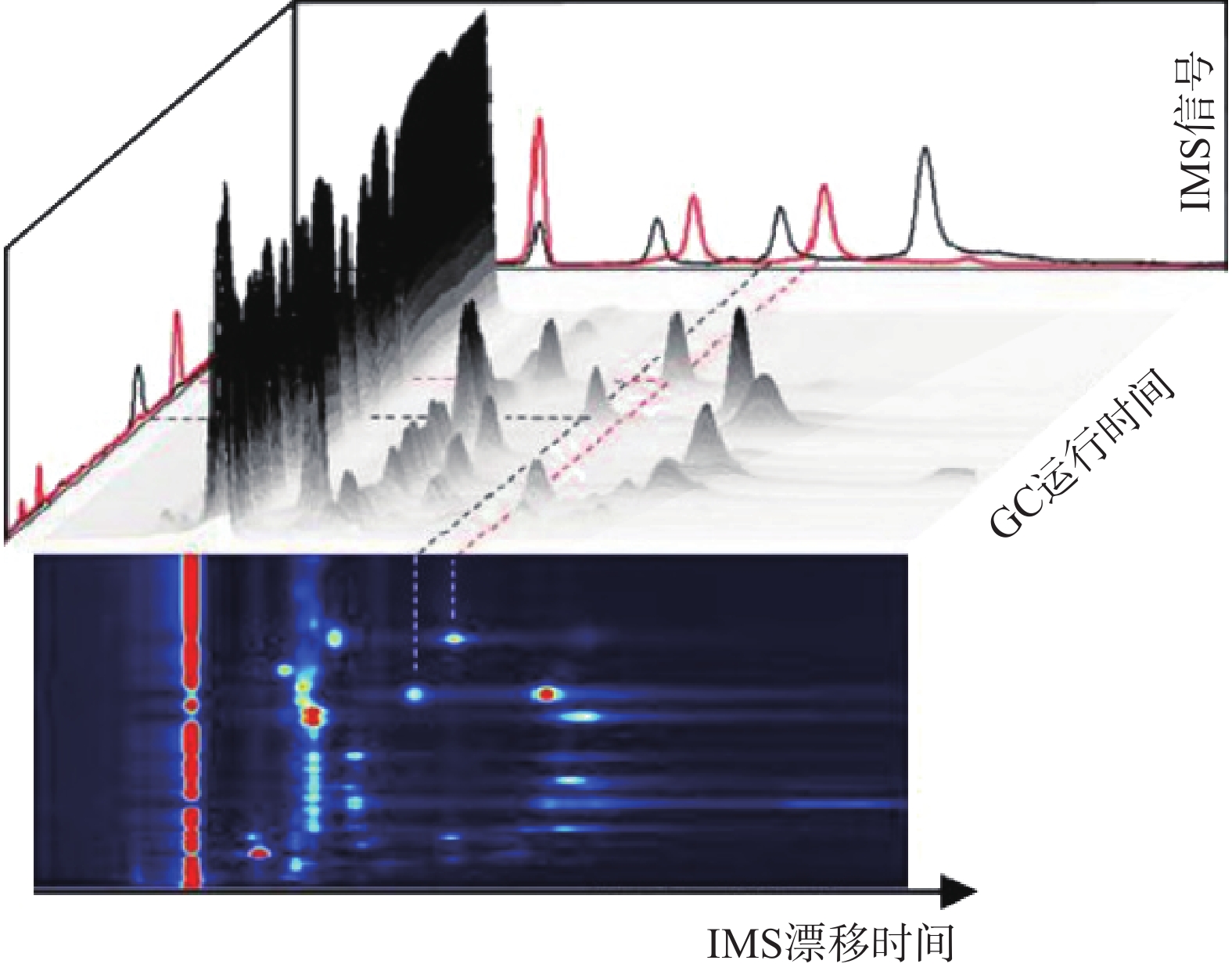
GC-IMS 技术的基本原理[2]是通过将样品混合物引进气相色谱仪进行分离,样品分子和载气分子在离子源放射性物质的作用下发生一系列反应形成产物离子,这些产物离子在不同的电场驱动下通过离子门进入迁移区,与逆向而来的中性迁移气体分子发生碰撞而损失能量。产物离子在电场中的迁移速率不同,到达检测器上的时间不同,从而使样品差异化分离(如图1),最后可得到一个包含有离子迁移时间(X轴),气相色谱保留时间(Y轴),离子强度(Z轴)的三维谱图(如图2)。
顶空-气相色谱-离子迁移谱(HS-GC-IMS)是应用于检测药材中VOCs最为广泛的方式之一 [3]。其原理是首先将样品中的VOCs通过热孵育和振摇使之从药材中逸出,随后抽取顶空气体进行分析,避免复杂基质干扰,再使之进入气相色谱(GC)、离子迁移谱(IMS)中,得到相应谱图。HS-GC-IMS无需样品预处理,在分析复杂样品或需要快速检测场景方面更具优势。
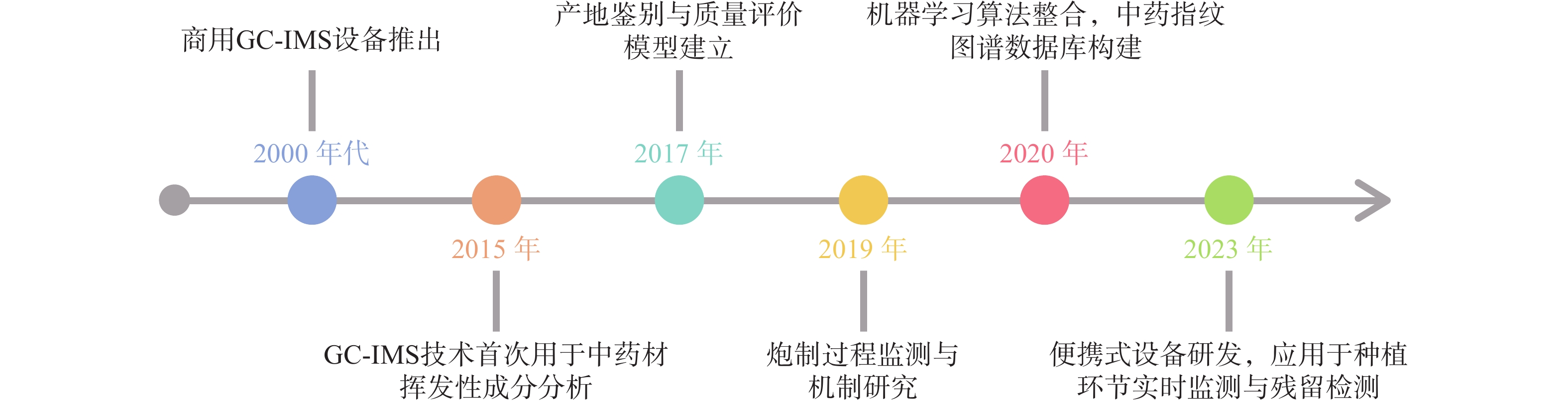
自从1972年第一张经过GC分离的IMS谱图出现[2],早期主要用于军事领域(如化学战剂检测)和毒品筛查的先进技术逐渐面向市场,应用于研究(如图3),引进国内后广泛使用于中药研究。现阶段,通过GC-IMS捕捉到中药材中的微量VOCs,将其应用于区分中药的不同种类和产地、监控中药炮制过程以及中药复方组分分析等[4],为中药材的产地及真伪鉴别提供可靠的依据,同时也为质量控制和药效研究等方面提供数据支持。
2. 在中药鉴定中的应用
2.1 同属不同种的中药鉴别
同属不同种之间的中药材亲缘关系比较接近,在VOCs种类差异上不显著,利用GC-IMS技术善于捕捉挥发性化合物种类微小差异的特点,可对样品进行品种鉴别。
陈皮中主要含有三萜类、挥发油类等几百种成分,有平喘止咳、调节血管等药理作用[5-6],刘主洁等[7]和Lv等[8]采用HS-GC-IMS对陈皮和广陈皮样品中VOCs进行研究,分析二者的特征信号,以邻氨基苯甲酸二甲酯为广陈皮最明显的特征标记物,柠檬烯、癸醛等也可作为广陈皮的特征信号,利用这些特征信号可对陈皮和广陈皮进行区分。山莓是覆盆子的混淆品之一,为保证用药安全有效,有必要对两者进行区分[9],严爱娟等[10]采用GC-IMS技术检测山莓和覆盆子VOCs,明确鉴定出覆盆子特征成分较山莓多,其中癸醛、1-辛烯-3-醇等在覆盆子样品中的含量更高,苯甲醛、2-丁酮等在山莓样品中的含量更高,以此来实现山莓和覆盆子的区分。彭旭阳等[11]采用GC-IMS分析新疆和田地区“梭梭”和“红柳”肉苁蓉这两种不同寄主肉苁蓉挥发性物质之间的不同,找到了二者主要差异物质为苯甲醛、庚醛等。
2.2 中药不同产地鉴别
道地药材是指生长在特定的自然生态环境中,经过一系列技术培育和加工而成,且被公认为比其他地方生产的同种药材的质量和治疗效果好的药材[12]。但随着产地的迁移、品种的引入,在外观性状上对道地药材和其他产地的中药进行简单鉴别已经不能满足需求。
王振洲等[13]采用GC-IMS对来自不同产区的人参VOCs进行检测,发现2,5-二甲基吡嗪和2,6-二甲基吡嗪等VOCs在吉林集安四年生人参中含量较高,而吉林敦化四年生人参中含量相对较低,由此鉴别四年生吉林集安和敦化的人参。西洋参同属于五加科人参属,成功引种进入我国后,辽宁、吉林和山东是其主产区[14],王燕等[15]使用GC-IMS技术对美国、加拿大、山东等五个不同产地的西洋参进行研究,共鉴定出53种VOCs,其中美国西洋参中2-庚酮等成分的含量较高,而加拿大产α-蒎烯等成分的含量显著高于其它产区,中国产芳樟醇等成分的含量较高,其中吉林产地的含量是辽宁、山东产地的2.60和3.60倍,通过这些显著差异可对西洋参进行产地溯源和鉴别。
李曼祎等[16]使用GC-IMS技术对新疆、宁夏、内蒙古、青海四个主要产区的枸杞进行化学成分测定,分析出了四个地区含量差异较大的16种物质,发现内蒙古枸杞区别于另外三个产地特有的物质为正丁醇,同时筛选出了叶醇等五种标记性物质对枸杞产地进行区分。Li[17]等分别采用HS-SPME-GC-MS和HS-GC-IMS检测河北、河南、江苏、浙江、安徽以及山东6个不同地区的五味子,结果显示安徽的五味子萜类物质含量较高,同时该研究也证明了HS-GC-IMS对样品的分类效果优于HS-SPME-GC-MS。山东为瓜萎的道地产区[18],河北也是瓜萎的重要产区,不同产地的瓜萎可能在品质及所含成分上有所差别,从而对药效也会产生一定的影响[19-20]。张敏敏等[19]采用GC-IMS在瓜萎皮样品中共鉴定出醛类和醇类等88种VOCs,分析发现山东瓜蒌皮中2-庚酮、正壬醛等物质的含量低于河北瓜蒌皮,但1-己醇、糠醇等含量更高的结论,通过这些差异基本实现了两地区瓜萎皮的区分。Li等[21]采用HS-GC-IMS联合PCA建立松茸特征图谱,对分别来自云南和四川的松茸样品以及它们的菌盖和菌柄进行研究,发现虽然指纹图谱相似度较高,但各自也有其特征性挥发物:苯乙醛和糠醇等仅在云南产松茸的菌柄中被发现,戊烷仅在四川产松茸的菌柄中被发现,戊醛仅在云南产松茸的菌盖中检出,甲基吡嗪仅在四川产松茸的菌盖中检出,通过特征性挥发物的不同,可对分别产自云南和四川的松茸进行区分。
2.3 中药复方中主成分的鉴别
中药复方成分众多且复杂,确保其质量符合标准、疗效可靠、使用安全是关键,采用HPLC特征图谱进行分析是目前公认的良好方法之一[22]。GC-IMS检测灵敏度高、分离效果好,是对HPLC表征中药复方质量分析的有益补充。
Yuan等[23]设置了对照组、慢性不可预测轻度应激(CUMS)组和CUMS+百合鸡子黄汤组,采用HS-GC-IMS等方法对百合鸡子黄汤治疗CUMS大鼠粪便中挥发性化合物含量进行研究,鉴定出了11个生物标志物,找出了对照组大鼠粪便样品中甲硫醚含量较高,而CUMS组则较低,同时百合子鸡汤组抑郁表现出保护性干预作用;Yin等[24]采用HS-GC-IMS对开心散中的挥发性化合物进行分析,鉴别出β-细辛酮等十种VOCs可作为开心散的质量标记物,进一步为开心散质量控制及药效机制等的相关研究奠定基础;李巍等[25]利用HS-GC-IMS对清感秋饮中的VOCs进行定性定量分析,共鉴定出120种VOCs,其中,紫苏属酮、β-石竹烯等可能为其主要药效成分。
3. 在中药加工炮制中的应用
中药的加工炮制是提高临床疗效的重要手段,也是保证临床用药安全的重要措施[26]。应用不同的炮制方法可能会引起中药中的化学成分发生含量加减、成分转化与破坏等变化,采用GC-IMS技术对中药加工炮制过程中的VOCs进行动态监控,对于炮制工艺的规范、制定更优炮制方案等具有指导意义。
干燥是中药材和饮片加工制备过程中的重要且关键的环节之一,而中药中的VOCs易受干燥工艺的影响,对于富含VOCs的中药,准确控制干燥工艺有利于减少有效成分的损失。陈树鹏等[27]采用GC-IMS等技术确定烘干样品的整体香气属性优于晒干样品,这可能是由于烘干工艺对环境温度的调整使得烘干过程更有利于果香、柑橘香以及甜香香气保留。其中苯乙酸乙酯、乙酸乙酯等 12 种物质为晒干主要成分,2-甲基-1-丁醇、(E)-2-己烯-1-醇等为烘干主要成分;Wang等[24]在8S-GC-IMS技术的辅助下,了解了柑橘皮干燥以及不同条件下挥发性成分情况,研究柑橘皮各成分在不同干燥温度下的缺失,其中,70 ℃下干燥会导致2,2-苯基-1-苦基肼基和铁还原抗氧化能力显著降低。Yu等[29]通过GC-MS比较总离子色谱图中的峰面积与苯乙酸乙酯的峰面积,将VOCs的含量进行半定量再通过GC-IMS呈现图谱对结果进行比较,确定VOCs的身份,从而证实晒干有利于两个品种的网纹柑橘中萜醇类化合物的保存,热风干燥有利于脂肪族醛和倍半萜的保存,而冷冻干燥是保存酯类和酚类物质的最佳方法;Zhou等[30]对肉苁蓉进行酒制增效后粉碎、超微粉碎、醇提、水提等处理,采用HS-GC-IMS方法检测其VOCs并建立指纹图谱,发现增效处理的肉苁蓉VOCs的种类和含量有所减少,分析原因可能为各种化学物质之间在处理过程中会发生化学变化,而新鲜肉苁蓉则保存了更多种类的VOCs,超微粉碎处理和水提处理后的肉苁蓉挥发性化合物主要以醛类为主。除此之外,将其它中药基于GC-IMS技术在不同炮制方法中的应用汇总于表1。
表 1 GC-IMS技术在炮制研究方面的应用作者 药材及炮制方法 采用方式 实例 高以丹等 [31] 柴达木枸杞
冷冻干燥、自然阴干、热风烘干GC-IMS 从枸杞样品中鉴定出反-2-壬烯醛、2,4-庚二烯醛等52种VOCs,表明冷冻干燥法比自然阴干、热风烘干以及微波干燥更好,能够有效保留枸杞中的VOCs,使枸杞保持较高的品质。 时海燕等 [32] 六神曲
生品、炒品、焦品HS-GC-IMS 从六神曲生品、炒品和焦品中鉴别出60种化合物通过比较种类和差异,得出炒神曲比焦神曲健胃消食的效果更好。 林秀敏等 [33] 当归
酒洗、酒炙、酒浸GC-IMS 2-十一烯醛、丙酮等为酒洗与酒浸当归的主要差异性物质,2-十一烯醛、丙酮等为酒洗与酒炙当归的主要差异性物质,2-十一烯醛、辛酸乙酯等为酒浸与酒炙当归的主要差异性物质。 武旭等 [34] 胆南星
发酵炮制GC-IMS 发酵炮制有助于胆南星矫味矫臭 王雨晨等 [35] 太子参
常温晾干、晒干、热风干燥、
真空冷冻干燥GC-IMS 40 ℃热风干燥可以有效保留太子参样品中的VOCs,与晒干、晾干样品无差异,但真空冷冻干燥对太子参挥发性成分的影响较大,会造成挥发性成分以及风味的损失 焦焕然等 [36] 侧柏叶
常温晾干、晒干,热风干燥、
变温干燥GC-IMS 40 ℃和60 ℃热风干燥能够较好地保留瓜蒌样品中的核苷类和黄酮类成分 4. 与电子鼻联用
国内外也有许多采用GC-IMS与电子鼻联用对中药挥发性成分进行研究。电子鼻是一种通过模拟人嗅觉系统对检测物质进行品质评价的感官仪器,其原理是通过传感器阵列对气味分子进行检测和响应,将产生的信号经过预处理后送入模式识别系统,通过指纹图谱对挥发性成分或是气体进行定量或定性分析[37]。两种技术的联用为实验的结果研究提供了更高的准确度。
Feng等 [38]采用GC-IMS、GC-MS对不同地理标志的八种花椒的VOCs进行测定,证明了两种方法均可用于对不同花椒的分类,但相较之下GC-IMS操作时间更短,且有能够检测到含量很低物质,结果表明红花椒比青花椒能够释放出更多的萜烯、酯类和更少的醇类,同时该研究还与电子鼻联用表征花椒中的香气物质,W1W、W2W和W5S传感器对花椒样品VOCs的响应更强,说明花椒产品中可能含有更高丰度的萜烯、有机硫化物和氮氧化物。陈小爱等 [39]利用GC-MS、GC-IMS和电子鼻技术联用,分析老香黄在发酵期间的VOCs变化,GC-MS共鉴别出包括醇类等八个种类的46种VOCs,GC-IMS则检测出包括杂环类等九个类别的38种VOCs,同时电子鼻PCA有效区分了不同发酵时间的样品,发现发酵6个月后老香黄挥发性组分开始发生较大的变化,其中柠檬烯等14种是发酵期间含量较高且相对稳定的成分,发酵过程中产生的庚醛、糠醛等是构成老香黄特有气味的特征性成分。王世丽等 [40]通过电子鼻辨识南北柴胡气味特征物质与GC-IMS检测其挥发性成分,发现南北柴胡中短链烷烃、醛类等物质差异较大,癸醛、异戊烯醛等可作为南柴胡的特征物质,2-甲基丙酸、3-甲基丁醇可作为北柴胡的特征物质,此外乙酸、乙酸甲酯等成分在北柴胡中显著高于南柴胡。
5. 总结和展望
GC-IMS在中药研究中的应用前景非常广阔,不仅可以对同属不同种、不同产地来源、不同采收期以及不同贮存时间的中药VOCs进行分析鉴别,还可以帮助分析炮制前后中药VOCs含量变化以及在复方中寻找质量标志物,为药物质量控制与药效研究提供帮助。另外,随着技术的不断进步和中药现代化需求的增加,GC-IMS可以与特征图谱相结合,构建特征指纹图谱;也可以与电子鼻等其他分析手段融合,发挥出新的效果,让其所能提供的信息更加全面。但该项技术作为新兴科技仍需解决许多问题,比如应探索融合数据库的体系架构[41]。目前,GC-IMS通常使用的数据库为NIST出版的标准质谱图,对于中药VOCs的专业数据库搭建还不全面,部分VOCs需要自行判断建立文档保存入库,对实验进程造成不便。由于中药挥发性成分复杂,GC-IMS可能因峰重叠导致部分成分无法准确定性,例如分析复方丹参片时,GC-IMS仅能明确鉴定其中60%的化合物,需GC-MS辅助验证。而且GC-IMS对象单一,无法检测多糖、生物碱等非挥发性成分,难以全面评价中药质量。
总的来说,GC-IMS技术为中药研究提供了一种新的科学工具,有利于推动中药科学研究深入,也为中药产业的发展走向国际化和标准化提供支持。
-
[1] CHO N, SHAW J, KARURANGA S, et al. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045[J]. DIABETES RES CLIN PR, 2018, 138:271-281. doi: 10.1016/j.diabres.2018.02.023 [2] MARTÍNEZ-CASTELAO A, NAVARRO-GONZÁLEZ J, GÓRRIZ J, et al. The concept and the epidemiology of diabetic nephropathy have changed in recent years[J]. J Clin Med, 2015, 4(6):1207-1216. doi: 10.3390/jcm4061207 [3] SARAN R, ROBINSON B, ABBOTT K C, et al. US renal data system 2016 annual data report: epidemiology of kidney disease in the United States[J]. Am J Kidney Dis, 2017, 69(3):A4. doi: 10.1053/j.ajkd.2017.01.036 [4] VALENCIA WM, FLOREZ H.How to prevent the microvascular complications of type 2 diabetes beyond glucose control[J]. BMJ, 2017, 356: j1018. [5] CENTERS FOR DISEASE CONTROL AND PREVENTION (CDC). Incidence of end-stage renal disease attributed to diabetes among persons with diagnosed diabetes: - United States and Puerto Rico, 1996-2007[J]. MMWR Morb Mortal Wkly Rep, 2010, 59(42):1361-1366. [6] ZHANG L X, LONG J Y, JIANG W S, et al. Trends in chronic kidney disease in China[J]. N Engl J Med, 2016, 375(9):905-906. doi: 10.1056/NEJMc1602469 [7] OSAMA G, NASHWA F, NARAYANAN N, et al. Diabetic kidney disease: world wide difference of prevalence and risk factors[J]. J Nephropharmacology, 2016, 5(1):49-56. [8] STENVINKEL P. Chronic kidney disease: a public health priority and harbinger of premature cardiovascular disease[J]. Journal of internal medicine 2010; 268: 456-467. [9] IATCU C O, STEEN A, COVASA M. Gut microbiota and complications of type-2 diabetes[J]. Nutrients, 2021, 14(1):166. doi: 10.3390/nu14010166 [10] WILSON TANG W H, KITAI T, HAZEN S L. Gut microbiota in cardiovascular health and disease[J]. Circ Res, 2017, 120(7):1183-1196. doi: 10.1161/CIRCRESAHA.117.309715 [11] SCHROEDER B O, BÄCKHED F. Signals from the gut microbiota to distant organs in physiology and disease[J]. Nat Med, 2016, 22(10):1079-1089. doi: 10.1038/nm.4185 [12] YANG G, WEI J, LIU P, et al. Role of the gut microbiota in type 2 diabetes and related diseases[J]. METABOLISM, 2021, 117:154712. doi: 10.1016/j.metabol.2021.154712 [13] SHARMA M, LI Y Y, STOLL M L, et al. The epigenetic connection between the gut microbiome in obesity and diabetes[J]. Front Genet, 2020, 10:1329. doi: 10.3389/fgene.2019.01329 [14] JAWORSKA K, KOPACZ W, KOPER M, et al. Enalapril diminishes the diabetes-induced changes in intestinal morphology, intestinal RAS and blood SCFA concentration in rats[J]. Int J Mol Sci, 2022, 23(11):6060. doi: 10.3390/ijms23116060 [15] DU X, LIU J, XUE Y, et al. Alteration of gut microbial profile in patients with diabetic nephropathy[J]. Endocrine, 2021, 73(1):71-84. doi: 10.1007/s12020-021-02721-1 [16] QIN J J, LI Y R, CAI Z M, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes[J]. Nature, 2012, 490(7418):55-60. doi: 10.1038/nature11450 [17] KARLSSON F H, TREMAROLI V, NOOKAEW I, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control[J]. Nature, 2013, 498(7452):99-103. doi: 10.1038/nature12198 [18] ZHANG L, LU Q Y, WU H, et al. The intestinal microbiota composition in early and late stages of diabetic kidney disease[J]. Microbiol Spectr, 2023, 11(4):e0038223. doi: 10.1128/spectrum.00382-23 [19] KALANTAR-ZADEH K, JAFAR T H, NITSCH D, et al. Chronic kidney disease[J]. Lancet, 2021, 398(10302):786-802. doi: 10.1016/S0140-6736(21)00519-5 [20] WANG X, YANG S, LI S, et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents[J]. Gut, 2020, 69(12):2131-2142. doi: 10.1136/gutjnl-2019-319766 [21] WANG S L, SHAO B Z, ZHAO S B, et al. Impact of paneth cell autophagy on inflammatory bowel disease[J]. Front Immunol, 2018, 9:693. doi: 10.3389/fimmu.2018.00693 [22] BUCKLEY A, TURNER J R. Cell biology of tight junction barrier regulation and mucosal disease[J]. Cold Spring Harb Perspect Biol, 2018, 10(1):a029314. doi: 10.1101/cshperspect.a029314 [23] MAHMOODPOOR F, RAHBAR SAADAT Y, BARZEGARI A, et al. The impact of gut microbiota on kidney function and pathogenesis[J]. Biomed Pharmacother, 2017, 93:412-419. doi: 10.1016/j.biopha.2017.06.066 [24] VAZIRI N D, YUAN J, NORRIS K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease[J]. Am J Nephrol, 2013, 37(1):1-6. doi: 10.1159/000345969 [25] MATHEWSON N D, JENQ R, MATHEW A V, et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease[J]. Nat Immunol, 2016, 17(5):505-513. doi: 10.1038/ni.3400 [26] HUANG X Y, OSHIMA T, TOMITA T, et al. Butyrate alleviates cytokine-induced barrier dysfunction by modifying claudin-2 levels[J]. Biology, 2021, 10(3):205. doi: 10.3390/biology10030205 [27] NOWARSKI R, JACKSON R, GAGLIANI N, et al. Epithelial IL-18 equilibrium controls barrier function in colitis[J]. Cell, 2015, 163(6):1444-1456. doi: 10.1016/j.cell.2015.10.072 [28] TONG L C, WANG Y, WANG Z B, et al. Propionate ameliorates dextran sodium sulfate-induced colitis by improving intestinal barrier function and reducing inflammation and oxidative stress[J]. Front Pharmacol, 2016, 7:253. [29] FENG Y H, WANG Y, WANG P, et al. Short-chain fatty acids manifest stimulative and protective effects on intestinal barrier function through the inhibition of NLRP3 inflammasome and autophagy[J]. Cell Physiol Biochem, 2018, 49(1):190-205. doi: 10.1159/000492853 [30] SAYIN S, WAHLSTRÖM A, FELIN J, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist[J]. Cell Metab, 2013, 17(2):225-235. doi: 10.1016/j.cmet.2013.01.003 [31] SCHAAP F G, TRAUNER M, JANSEN P L M. Bile acid receptors as targets for drug development[J]. Nat Rev Gastroenterol Hepatol, 2014, 11(1):55-67. doi: 10.1038/nrgastro.2013.151 [32] HUANG W D, MA K, ZHANG J, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration[J]. Science, 2006, 312(5771):233-236. doi: 10.1126/science.1121435 [33] WANG X X, WANG D, LUO Y H, et al. FXR/TGR5 dual agonist prevents progression of nephropathy in diabetes and obesity[J]. J Am Soc Nephrol, 2018, 29(1):118-137. doi: 10.1681/ASN.2017020222 [34] XIAO H M, SUN X H, LIU R B, et al. Gentiopicroside activates the bile acid receptor Gpbar1 (TGR5) to repress NF-kappaB pathway and ameliorate diabetic nephropathy[J]. Pharmacol Res, 2020, 151:104559. doi: 10.1016/j.phrs.2019.104559 [35] MARQUARDT A, AL-DABET M M, GHOSH S, et al. Farnesoid X receptor agonism protects against diabetic tubulopathy: potential add-on therapy for diabetic nephropathy[J]. J Am Soc Nephrol, 2017, 28(11):3182-3189. doi: 10.1681/ASN.2016101123 [36] DU Y, YANG Y T, TANG G, et al. Butyrate alleviates diabetic kidney disease by mediating the miR-7a-5p/P311/TGF-β1 pathway[J]. FASEB J, 2020, 34(8):10462-10475. doi: 10.1096/fj.202000431R [37] DONG W P, JIA Y, LIU X X, et al. Sodium butyrate activates NRF2 to ameliorate diabetic nephropathy possibly via inhibition of HDAC[J]. J Endocrinol, 2017, 232(1):71-83. doi: 10.1530/JOE-16-0322 [38] XU Y H, GAO C L, GUO H L, et al. Sodium butyrate supplementation ameliorates diabetic inflammation in db/db mice[J]. J Endocrinol, 2018, 238(3): 231-244. [39] RAMEZANI A, MASSY Z A, MEIJERS B, et al. Role of the gut microbiome in uremia: a potential therapeutic target[J]. Am J Kidney Dis, 2016, 67(3):483-498. [40] ZHANG F, QI L L, FENG Q Y, et al. HIPK2 phosphorylates HDAC3 for NF-κB acetylation to ameliorate colitis-associated colorectal carcinoma and sepsis[J]. Proc Natl Acad Sci USA, 2021, 118(28):e2021798118. doi: 10.1073/pnas.2021798118 [41] HU X Y, LI S M, FU Y H, et al. Targeting gut microbiota as a possible therapy for mastitis[J]. Eur J Clin Microbiol Infect Dis, 2019, 38(8):1409-1423. doi: 10.1007/s10096-019-03549-4 [42] HU Z B, LU J, CHEN P P, et al. Dysbiosis of intestinal microbiota mediates tubulointerstitial injury in diabetic nephropathy via the disruption of cholesterol homeostasis[J]. Theranostics, 2020, 10(6):2803-2816. doi: 10.7150/thno.40571 [43] LU C C, HU Z B, WANG R, et al. Gut microbiota dysbiosis-induced activation of the intrarenal renin-angiotensin system is involved in kidney injuries in rat diabetic nephropathy[J]. Acta Pharmacol Sin, 2020, 41(8):1111-1118. doi: 10.1038/s41401-019-0326-5 [44] GRUPPEN E G, GARCIA E, CONNELLY M A, et al. TMAO is associated with mortality: impact of modestly impaired renal function[J]. Sci Rep, 2017, 7(1):13781. doi: 10.1038/s41598-017-13739-9 [45] SUN G P, YIN Z M, LIU N Q, et al. Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity[J]. Biochem Biophys Res Commun, 2017, 493(2):964-970. doi: 10.1016/j.bbrc.2017.09.108 [46] AL-OBAIDE M, SINGH R, DATTA P, et al. Gut microbiota-dependent trimethylamine-N-oxide and serum biomarkers in patients with T2DM and advanced CKD[J]. J Clin Med, 2017, 6(9):86. doi: 10.3390/jcm6090086 [47] YANG M X, ZHANG R, ZHUANG C F, et al. Serum trimethylamine N-oxide and the diversity of the intestinal microbial flora in type 2 diabetes complicated by diabetic kidney disease[J]. Clin Lab, 2022, 68(5): 10.7754/Clin. Lab. 2021.210836. [48] FANG Q, ZHENG B J, LIU N, et al. Trimethylamine N-oxide exacerbates renal inflammation and fibrosis in rats with diabetic kidney disease[J]. Front Physiol, 2021, 12:682482. doi: 10.3389/fphys.2021.682482 [49] MAO Z H, GAO Z X, LIU D W, et al. Gut microbiota and its metabolites–molecular mechanisms and management strategies in diabetic kidney disease[J]. Front Immunol, 2023, 14:1124704. doi: 10.3389/fimmu.2023.1124704 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 6856
- HTML全文浏览量: 2840
- PDF下载量: 47
- 被引次数: 0
 下载:
下载:
